




Abstract
The existence of antibodies to myelin oligodendrocyte glycoprotein (MOG) in some patients with CNS demyelinating disease has been recognised for 30 years, but their clinical utility as biomarkers, and potential pathogenicity in humans has only become apparent in the past 15 years. The advent of more precise live cell-based assays for antibody detection in serum and cerebrospinal fluid (CSF) has greatly refined the clinical phenrotype of demyelinating diseases associated with MOG antibodies. Distinct patterns of MOG antibody associated disorder (MOGAD) include acute disseminated encephalomyelitis (ADEM) in children; and overlap with neuromyelitis optica spectrum disorders (including classical Devic’s presentations), optic neuritis, transverse myelitis, and focal encephalitis in both children and adults. A number of other rare presentations have also been described. Here we summarise what is currently known of the pathophysiology, clinical presentation and management of MOGAD.
Introduction
Myelin Oligodendrocyte Glycoprotein (MOG) antibody associated disorder (MOGAD) is a unique demyelinating pathology affecting adult and paediatric populations. Its clinical presentation, investigation, management and prognostic considerations distinguish it from multiple sclerosis (MS) and AQP4 antibody associated neuromyelitis optica spectrum disorder (AQP4-NMOSD).1 Recognition of this distinction promotes targeted therapy and prospective research.
Pathogenesis
MOG is expressed on the outer sheath of myelin by mammalian CNS oligodendrocytes, prominently exposed to the extracellular space. While its biological role is poorly understood, its association with CNS disease is increasingly apparent.2 MOG antibody is encephalitogenic in vitro. It is likely to be peripherally produced, frequently of the IgG1 isotype, and demonstrates reactivity to an extracellular antigenic region around Proline42 in a large subset of patients.3 How it bypasses the blood-brain barrier remains unknown. MOGAD-like pathology has been induced by encephalitic viruses in transgenic mice expressing MOG antibody.2 Pathologically in the brain MOGAD, like MS, shows white matter demyelination characterised by perivenous inflammatory aggregates. In MS, lesions are predominantly leukocortical, have a CD8 T-cell infiltrate with chronic radial expansion, whilst in MOGAD plaques are intracortical, associated with a CD4 T-cell inflammatory infiltrate and show preserved AQP4 expression.4
Epidemiology
The prevalence of MOGAD is not well established. A recent comparative survey has shown MOGAD is at least twice as common as AQP4-NMOSD.5 Existing cohort studies are small and frequently combine paediatric and adult samples. Age of onset ranges from 0–90 years. Unlike AQP4-NMOSD and MS, a female preponderance is not evident. There is insufficient data to establish ethnicity or latitude as risk factors. There are limited reports of association with other autoimmune diseases but fewer than seen with AQP4-NMOSD.2,6
Clinical presentation
The MOGAD clinical phenotype is diverse and varies with age.2,7 A prodromal infection is reported in up to 47% in some studies.6
Optic neuritis (ON) is the most frequent presentation in adults and children over 9 years. The rate of simultaneous or sequential, bilateral ON is higher than seen in MS or AQP4-NMOSD.7 Papillitis is frequently identified in MOGAD, consistent with the anterior visual pathway involvement often seen in this condition.6,8 Symptom resolution is anticipated but up to 20% of patients may have permanent visual impairment. Relapse of ON is common.
Transverse myelitis (TM) may occur in isolation or with ON to mimic Devic’s disease. It is the second most frequent presentation in adults and older children.2 TM in MOGAD may mimic that seen in AQP4-NMOSD and MS. Both longitudinally extensive and short segment TM are observed. Early bowel, bladder and sexual dysfunction, reflecting central cord and conus involvement, increases suspicion for MOGAD.8,9 Residual paraparesis is less common than in AQP4-NMOSD.7
Acute disseminated encephalomyelitis (ADEM) is the most common initial presentation in children under 9 years.2 Indeed, MOG antibody accounts for the majority of ADEM and non-ADEM autoimmune encephalitis (AE) presentations in children8,10 and is associated with a higher risk of relapse than MOG antibody negative disease.2
Paediatric MOGAD encephalitis can result in poor long term outcome (mRS>2) with progression to leukodystrophy or cortical atrophy.10 ADEM in adults is rare but MOG antibody is increasingly identified in clinically diverse cases of focal and rhombo- encephalitis, with and without leptomeningeal enhancement.7,8 Seizures and refractory status epilepticus have been reported in both adults and children with cortical encephalitis or as an isolated phenomenon.10 Brain stem syndromes are recognised, but infrequent. The area postrema is less frequently affected than in AQP4-NMOSD.8 Patients with AE (including NMDAR and LGI1 AE) have been identified as having anti-MOG antibodies with and without current or prior clinical or MRI evidence of demyelination.11
Relapse rates in children and adults with MOGAD approach 30 and 40-84% respectively at 5 years, with the majority of second attacks occurring in the first 12 months.10,12,13 Delayed diagnosis and heterogeneous treatment may have inflated these figures. Prospective trials examining relapse following targeted immunosuppression from diagnosis in MOGAD will increase accuracy of rates. Risk of relapse is unrelated to clinical phenotype, may differ from initial presentation in both phenotype and severity, is associated with cumulative disability, and can be associated with rapid weaning and cessation of steroid treatment.2,6,12
Investigation
Given important differences in management between these three demyelinating aetiologies, prompt diagnosis is paramount. All patients with monophasic or relapsing ON, TM or ADEM, without typical MS brain lesions, should be tested for both MOG and AQP4 antibodies before diagnosis and treatment of MS is considered. Screening patients with typical radiological features of MS for MOG antibodies is not recommended.15 Testing for MOG antibody should be reserved for circumstances of high pre-test probability.1,8 According to international expert consensus these include the above clinical syndromes with radiological evidence of demyelination and at least one associated feature described in Table 1.1
Table 1. Proposed diagnostic criteria for MOGAD
| Clinical Syndrome | Supportive Features | |
|---|---|---|
| Clinical | Radiological | |
| Optic Neuritis | Simultaneous bilateral disease Frequent relapse/recurrence Severe visual deficit Papillitis | Lesion longitudinally extensive and/or anterior to chiasm Perioptic GAD enhancement Absent supratentorial WM lesions |
| Transverse Myelitis | Severe/frequent episodes Residual sphincter/erectile dysfunction | Lesion/atrophy longitudinally extensive and/or in conus Central canal pseudodilatation and/or pencil thin GAD enhancement H shape hyperintensity on axial sequence involving grey and WM Leptomeningeal GAD enhancement Absent supratentorial WM lesions |
| Encephalitis | Behavioural change Altered level of consciousness Seizures | ADEM appearance Thalamic and Basal Ganglia lesions Isolated brainstem lesions Cranial nerve root GAD enhancement Leptomeningeal GAD enhancement |
| Suggestive clinico-pathologic features across phenotypes |
|---|
| Recent history of infection/vaccination – Neutrophilic pleocytosis in CSF (paediatric cases) – No unmatched oligoclonal bands in CSF Steroid-dependent symptoms – Lack of response to MS specific therapy |
| GAD – gadolinium, WM – white matter |
Live cell-based assays against full length human MOG antigen performed on serum is the gold standard for MOG antibody detection. Though titres fluctuate with disease activity and treatment, their role in monitoring or prognosis has not been established1,3,7 A negative titre at 6 or 12 months may represent remission.8 However, clinical relapses have been reported after a transient seronegative period.7,13 Fixed assays, though more readily available, are less sensitive3 and have a 10-15% false negative rate.14 ELISA detection of MOG-IgG is unreliable.14 MOG antibody titre in CSF is lower than serum due to predominant extrathecal production.1 CSF may show isolated, elevated protein, neutrophilic pleocytosis, non-specific leucocytosis and unmatched oligoclonal bands in up to 30% of patients.
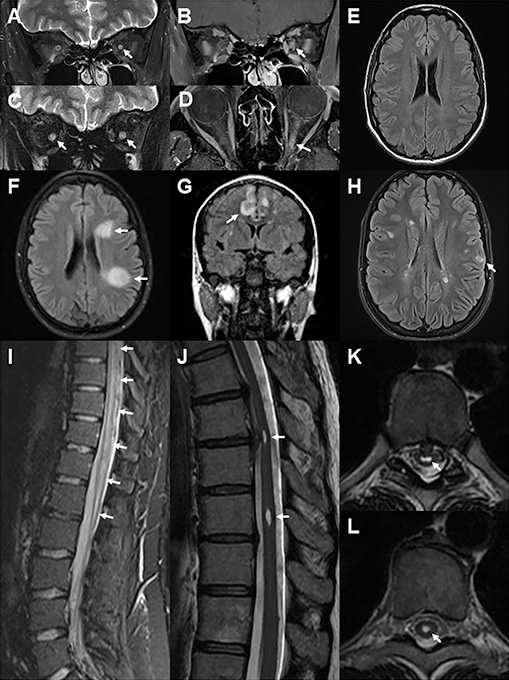
Attempts to define MRI changes distinct to MOGAD continue. Salient examples are provided in Figure 1. Bilateral longitudinally extensive involvement of the optic nerves is more commonly seen in MOGAD and AQP4-NMOSD compared to MS. Involvement of the intra-orbital and anterior segment of the optic nerve with relative sparing of the chiasm and retrochiasmatic tracts distinguishes MOGAD from AQP4-NMOSD (which more often affects the posterior segment of the optic nerve, chiasm, and optic tracts) and MS (limited segment).9 Optic perineuritis with perineural and orbital fat contrast enhancement is present in ~30% of cases.9 At follow-up, enhancement resolves, T2 high signal may persist and atrophy may be evident.9
{kind=link}
In TM, heterogeneous T2 high signal tends to involve both grey and white matter, >50% medullary cord and is associated with swelling or “pseudo-dilatation” of the ependymal canal.9 Signal is often brightest at the central part of the lesion. Enhancement can be variegated. Lesions therefore appear “cloud-like” on both T2 and contrast enhanced T1 sequences. Case reports document nodular or meningeal enhancement.9 At follow-up, enhancement resolves, T2 high signal may persist and atrophy may be evident but is less pronounced than in AQP4-NMOSD.
Brain lesions are only present in about a third of all cases and may be asymptomatic. Lesions may show ADEM-like confluence and a “fluffy” appearance even in adults (often without the encephalopathy seen in children presenting with ADEM) or have a more focal cortical appearance. Cloud-like infratentorial or brainstem lesions are rare but distinctive.9 The presence of Dawson’s fingers, lesions in the subcortical U fibres, adjacent to the lateral ventricles, or in the inferior temporal lobes are helpful in distinguishing MS from MOGAD.15
Management
Early recognition and treatment with high dose corticosteroids are essential to minimise morbidity. Patients frequently respond rapidly to steroid therapy. Plasma exchange has also been reported as an effective acute therapy.2 Relapses tend to be observed when prednisolone dose falls below 10mg/day, when tapering takes place over less than 2 months, or within the first few months following cessation.2,6 Prolonged corticosteroid therapy with a slow taper (over 6-9 months) is increasingly accepted as first line treatment following first presentation.2,6,8 A prospective analysis of treatment in patients with MOGAD showed one third relapsed despite B cell depletion.16 Retrospective studies of rituximab, azathioprine, mycophenolate, IVIg and corticosteroids demonstrate reductions in relapse rate, better EDSS at follow-up, and that maintenance immunosuppression should be instituted in patients with a relapsing disease course.2,6,10,12 Treatment with cyclophosphamide, methotrexate and mitoxantrone was too infrequent to extrapolate therapeutic efficacy.12 In the absence of treatment protocols, switching maintenance therapy in the event of relapse may be beneficial6. MS specific therapies have been largely shown to be less efficacious in MOGAD.
There are no established means to identify the small proportion of adult patients with monophasic MOGAD. The risk versus benefit of ceasing therapy needs to be discussed on a case by case basis. Given relapse seems to be more common in adult ON,3,6 the authors are reluctant to discontinue maintenance therapy in patients who have sustained permanent visual impairment. Patients with preserved visual acuity may have retinal nerve fibre layer atrophy, and OCT may be a useful adjunctive clinical investigation to help guide therapeutic decisions.
Prognosis
The long-term implications of MOGAD remain poorly understood. Follow-up studies have been short (3-5 years) and heterogenous. Morbidity correlates with the severity of attacks. The impact on visual acuity, mobility and cognition over the longer term may be less severe than AQP4-NMOSD or MS groups when effective treatment is initiated promptly. However, residual disability remains significant for many.2,6,12 Paediatric MOGAD has less relapse and better long term recovery than adult disease, so combined cohort studies may underestimate the likelihood of relapse and morbidity in adults.7,10
Conclusions
MOGAD is a distinct demyelinating pathology affecting adults and children. Clinical features are diverse and may mimic MS, AQP4-NMOSD or other autoimmune encephalitides. Prompt diagnosis relies on clinical suspicion, MRI findings and access to live cell-based assays. This is essential as early immunosuppressive treatment reduces morbidity, and maintenance immunosuppression in the event of a relapsing course prevents accumulated disability over time. Prospective research should continue to define the clinical spectrum and work toward evidence-based diagnostic and management guidelines. Separate consideration of paediatric and adult cohorts is essential.
References
1. Jarius S, Paul F, Aktas O, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation 2018;15(1):134. https://doi.org/10.1186/s12974-018-1144-2
2. Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol 2019;15(2):89-102. [published Online First: 2018/12/19] https://doi.org/10.1038/s41582-018-0112-x
3. Tea F, Lopez JA, Ramanathan S, et al. Characterization of the human myelin oligodendrocyte glycoprotein antibody response in demyelination. Acta Neuropathol Commun 2019;7(1):145. [published Online First: 2019/09/05] https://doi.org/10.1186/s40478-019-0786-3
4. Höftberger R, Guo Y, Flanagan EP, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathologica 2020 https://doi.org/10.1007/s00401-020-02132-y
5. O’Connell K, Hamilton-Shield A, Wood hall M, et al. Prevalence and incidence of neuromyelitis optica spectrum disorder, aquaporin-4 antibody-positive NMOSD and MOG antibody-positive disease in Oxfordshire, UK. J Neurol Neurosurg Psychiatry 2020;91(10):1126-28. published Online First: 2020/06/25] https://doi.org/10.1136/jnnp-2020-323158
6. Ramanathan S, Mohammad S, Tantsis E, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry 2018;89(2):127-37. https://doi.org/10.1136/jnnp-2017-316880
7. Cobo-Calvo A, Vukusic S, Marignier R. Clinical spectrum of central nervous system myelin oligodendrocyte glycoprotein autoimmunity in adults. Curr Opin Neurol 2019;32(3):459-66. [published Online First: 2019/02/15] https://doi.org/10.1097/WCO.0000000000000681
8. Jurynczyk M, Jacob A, Fujihara K, et al. Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations. Pract Neurol 2019;19(3):187-95. [published Online First: 2018/12/12] https://doi.org/10.1136/practneurol-2017-001787
9. Deneve M, Biotti D, Patsoura S, et al. MRI features of demyelinating disease associated with anti-MOG antibodies in adults. J Neuroradiol 2019;46(5):312-18. [published Online First: 2019/06/23] https://doi.org/10.1016/j.neurad.2019.06.001
10. Armangue T, Olive-Cirera G, Martinez-Hernandez E, et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol 2020;19(3):234-46. [published Online First: 2020/02/15] https://doi.org/10.1016/S1474-4422(19)30488-0
11. Martinez-Hernandez E, Guasp M, Garcia-Serra A, et al. Clinical significance of anti-NMDAR concurrent with glial or neuronal surface antibodies. Neurology 2020 [published Online First: 2020/03/13] https://doi.org/10.1212/WNL.0000000000009239
12. Cobo-Calvo A, Sepulveda M, Rollot F, et al. Evaluation of treatment response in adults with relapsing MOG-Ab-associated disease. J Neuroinflammation 2019;16(1):134. [published Online First: 2019/07/04] https://doi.org/10.1186/s12974-019-1525-1
13. Waters P, Fadda G, Woodhall M, et al. Serial Anti-Myelin Oligodendrocyte Glycoprotein Antibody Analyses and Outcomes in Children With Demyelinating Syndromes. JAMA Neurology 2020;77(1):82-93. https://doi.org/10.1001/jamaneurol.2019.2940
14. Reindl M, Schanda K, Woodhall M, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm 2020;7(2). https://doi.org/10.1212/NXI.0000000000000674 [published Online First: 2020/02/07
15. Jurynczyk M, Tackley G, Kong Y, et al. Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. J Neurol Neurosurg Psychiatry 2017;88(2):132-36. [published Online First: 2016/12/13] https://doi.org/10.1136/jnnp-2016-314005
16. Durozard P, Rico A, Boutiere C, et al. Comparison of the Response to Rituximab between Myelin Oligodendrocyte Glycoprotein and Aquaporin-4 Antibody Diseases. Ann Neurol 2020;87(2):256-66. [published Online First: 2019/11/15]
https://doi.org/10.1002/ana.25648
Key take-home messages
- MOGAD is a demyelinating pathology affecting adults and children, distinct from MS and NMOSD.
- Clinical features are diverse and may mimic MS, AQP4 positive NMOSD or other autoimmune encephalitides.
- Prompt diagnosis relies on clinical suspicion, MRI findings and access to a live cell-based assay.
- Early immunosuppressive treatment and maintenance therapy reduces morbidity and prevents relapse and accumulated disability over time.