


Abstract
Brain tumour classification provides a cornerstone for understanding tumour behaviour. The 2016 WHO classification of CNS tumours combines traditional histological grading with molecular genetics and introduces the concepts of ‘layered diagnosis’ and ‘integrated diagnosis’. An understanding of the basic concepts and important differences from the earlier WHO classifications is essential to guide clinical decision making. This paper aims to outline the key features of the 2016 WHO classification.
List of abbreviations:
IDH: Isocitrate Dehydrogenase TP53: Tumour protein (gene) 53 ATRX: Alpha-Thalassemia/ Mental retardation X-linked/ Transcriptional regulator TERT: Telomerase reverse transcriptase BRAF: proto-oncogene B-raf/ v-Raf murine sarcoma viral oncogene homolog B INI1: Integrase Interactor 1
Introduction
From Rudolf Virchow’s classification in 1863,1 through the seminal papers of Bailey and Cushing in 1926 explaining the relations between the developing brain and brain tumours,2 to the concept of tumour grading introduced in 1949 by James Kernohan and colleagues3 there have been many significant contributions in this field. Zulch et al published the first WHO classification in 1979, followed by the second (1993), third (2000) and fourth (2007) editions. A briefly popular alternative tumour grading system called the St. Anne – Mayo grading system was also published in 1988.4 Recently, partly as a result of The Cancer Genome Atlas (TCGA), our understanding of the molecular basis of brain tumours has grown exponentially. More precise molecular classification of brain tumours may improve the success rate of clinical trials by comparing similar molecular subtypes and lead to personalised therapeutic options for patients. The 2016 WHO classification introduced the concept of a “layered diagnosis”5 combining histology and molecular genetics. Layer 4 describes the molecular genetics, Layer 3 the grading, Layer 2 the histological features and Layer 1 being the final integrated diagnosis. This combined phenotypic and genotypic grouping puts tumours with similar prognostic markers together and guides treatment for biologically and genetically similar tumours. However, some disadvantages are inherent, like a delay in getting the final result due to the wait for molecular genetics testing, a potential change in the grade of the tumour with the final integrated diagnosis, and also, discrepancies in access to molecular facilities and expertise in some regions of the world. Relevant information regarding the commonest adult tumours are presented in this article, but the list is by no means exhaustive. It aims to provide clinicians a list of the conditions regularly encountered in a busy neurosurgical department.

There have been many changes introduced by the 2016 WHO classification, including loss of certain entities and introduction of new entities and variants based on a tumour’s combined histological and genetic characteristics.
Glioblastomas
Glioblastomas (GBMs) are highly malignant CNS tumours, with a reported incidence of 3.19 per 100000 person-years. With the best available treatment which includes maximal surgical resection and adjuvant radio and chemotherapy, the medial survival of patients stands at around 14 months from first diagnosis. With the understanding of the molecular basis of these tumours, many novel therapies aimed at the molecular structure of the tumours are being developed.
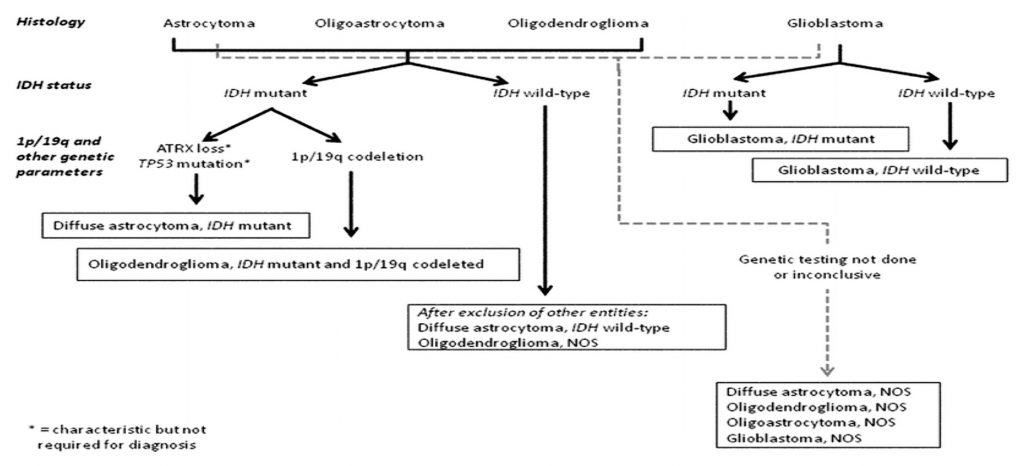
IDH mutation status subtypes
GBMs have now been classified based mainly on the IDH mutation status.7 IDH wild-type (lacking the mutation) and IDH-mutant are the 2 main newly defined entities. IDH wild-type GBM corresponds to the lesions previously described as primary glioblastoma and predominate in older patients whereas IDH mutant GBM, earlier known as secondary GBM, usually arises within a lower grade glioma and preferentially arises in younger patients.8
While IDH status is commonly assessed immunohistochemically using the R132H IDH1 antibody, patients who are 55 years and younger and negative by immunohistochemistry should ideally be sent for IDH1/2 sequencing to identify rarer IDH1/2 mutations.
The presence of IDH mutations is strongly predictive of a better outcome in secondary GBMs.9 Isocitrate dehydrogenases 1 and 2 (IDH1 and 2) are enzymes that convert isocitrate to a-ketoglutarate (aKG). Mutations of IDH1 and 2 bring about neomorphic activity, resulting in conversion of aKG to an oncometabolite called D-2-hydroxyglutarate (2-HG), the accumulation of which blocks cellular differentiation. This has led to the development of mutant IDH inhibitors (still in Phase 1 trials) which inhibit epigenetic dysregulation and induce cellular differentiation,10 leading to the hope of tailored treatments for patients with specific mutations.
Other GBM variants
A new variant called epithelioid glioblastoma has been added to the IDH wild-type category, joining others like giant cell glioblastoma and gliosarcoma. Typically seen in children and young adults, it harbours a BRAFV600E mutation in about 50% of cases.11 Another new addition to this category is GBM with primitive neuronal component (showing well demarcated nodules with primitive cells having neuronal differentiation, loss of GFAP expression, Homer-Wright rosettes, and an affinity for CSF seeding).12 Adenoid GBM mimics adenocarcinoma (pseudoepithelial pattern). Granular cell GBM shows presence of bland-looking granular cells typical of granular cell astrocytoma, along with features like frequent mitoses, pseudopalisading necrosis and endothelial hyperplasia. Historic entities like Gliomatosis cerebri and Primitive Neuroectodermal tumour (PNET) have been removed from the 2016 WHO classification. Gliomatosis is instead used to describe a pattern of spread of glioma.13 A category of glioblastoma NOS has been created for when full molecular evaluation cannot be performed.
Astrocytomas (Diffusely infiltrating astrocytomas)
Astrocytomas originate from immortalised astrocytes and have grades I – IV based on histological properties and presence of malignant features. The incidence is 5.4 per 100000 population. GBMs are grade IV astrocytomas. With optimal treatment, the prognosis for Grade I lesions (pilocytic astrocytomas, predominantly paediatric tumours) is >10 years, Grade II >5 years, and Grade IIIs 2-5 years.14
Astrocytomas are now defined by the presence or absence of IDH mutations and categorised as IDH mutant or IDH wild-type astrocytomas. If an R132 IDH1 immunohistochemistry assessment is negative, IDH sequencing should be performed in all cases. If determination of IDH status is not possible, the term NOS is allowed to be used. Hence, the terms now in use are:
• Diffuse astrocytoma, WHO grade II, IDH mutant (mut), wild-type (wt) and NOS (the term diffuse astrocytoma now encompassing earlier fibrillary and protoplasmic variants)
• Gemistocytic astrocytoma WHO grade II IDH mutant; and
• Anaplastic astrocytoma WHO grade III IDH mutant, wild-type and NOS. In nearly all cases of diffuse astrocytoma WHO grade II, IDHmut, there are TP53 mutations, and, in a majority, ATRX mutations can be found.15 Anaplastic astrocytomas exemplify the use of the layered diagnostic pathway; IDHwt are similar to GBM IDHwt in both genetic profile and prognosis and fare worse than GBM, IDHmut.13
Oligodendrogliomas
Oligodendrogliomas, according to the 2016 WHO classification, are defined as diffusely infiltrative gliomas with both IDH mutation and 1p/19q co-deletion. These can now include tumours that resemble astrocytomas phenotypically, but have the necessary genetic markers of IDH mutation and 1p/19q co-deletion.7 The presence of the 1p19q co-deletion improves overall survival in patients treated with radiotherapy and adjuvant chemotherapy comprising procarbazine, lomustine and vincristine.16 The incidence of oligodendrogliomas is calculated to be around 5-19% of all intracranial tumours and 25% of all gliomas. Median survival for Grade IIs is around 4-10 years and 3-4 years in Grade III lesions. Oligodendroglial tumours, NOS is a category used when it has not been possible to carry out molecular evaluation. The vast majority of oligodendrogliomas (along with IDH wild-type glioblastomas) harbour TERT mutations. The terms now in use in this category are:
• Oligodendroglioma WHO grade II, IDHmut & 1p19q co-deleted and NOS
• Anaplastic oligodendroglioma WHO grade III IDHmut & 1p19q co-deleted and NOS
• Oligoastrocytoma WHO grade II, NOS
• Anaplastic Oligoastrocytoma WHO grade III, NOS Oligoastrocytoma NOS is now only used for tumours which have both astrocytic and oligodendroglial phenotypes and there has been a failure or inability at establishing IDH/ 1p19q status. True oligoastrocytomas with dual genotypes (1p19q codeletion in oligodendroglial regions and ATRX loss or p53 expression in astrocytic regions)17 are extremely rare and are mostly resolved into either category using molecular evaluation.
Other Astrocytomas
This category includes:
• WHO grade I tumours like pilocytic astrocytoma and subependymal giant cell astrocytoma;
• WHO grade II pleomorphic xanthoastrocytomas and
• WHO grade III anaplastic pleomorphic xanthoastrocytomas (PXA III). Pilomyxoid astrocytomas were previously given a WHO grade II compared with pilocytic astrocytomas, but because of the genetic similarities between the two entities, it is uncertain whether the worse prognosis in pilomyxoid astrocytomas is related to their hypothalamic/ chiasmatic location. Therefore pilomyxoid astrocytomas are not assigned a WHO grade within the 2016 classification.7 Anaplastic pleomorphic xanthoastrocytomas WHO grade III are PXAs with 5 or more mitotic figures in 10 high power microscopic fields. Anaplastic PXAs WHO grade III have fewer BRAFV600E mutations than PXA grade IIs and have a worse prognosis.18
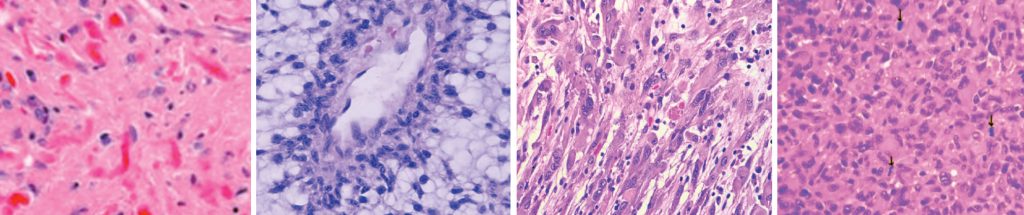
xanthoastrocytoma WHO grade II. Figure 14: Anaplastic pleomorphic Xanthoastrocytoma WHO grade III.
Meningiomas
Meningiomas are tumours thought to arise from arachnoidal cap cells in the CNS, accounting for 20% of all primary intracranial neoplasms, with an annual incidence of 2 per 100000 population.
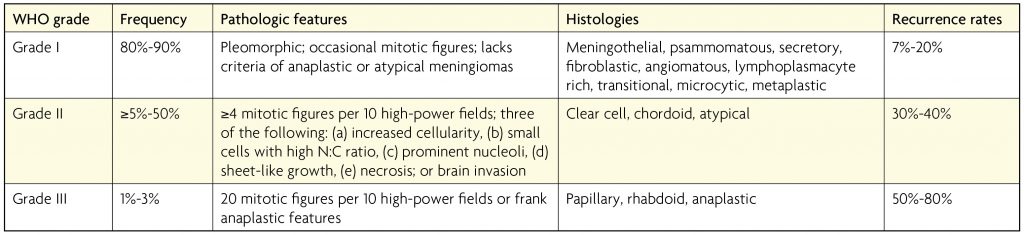
Meningioma classification is nearly unchanged from the previous editions, except for clarity regarding prognostic value of brain invasion in meningiomas, the presence of which categorises the meningioma very clearly as an atypical meningioma, WHO grade II.7 The category remains based upon histological appearance rather than molecular expression and contains the following WHO grade I variants: meningothelial, fibrous, transitional, psammomatous, angiomatous, microcystic, secretory, lymphoplasmacyte-rich, and metaplastic; WHO grade II variants: chordoid, clear cell and atypical; and the following WHO grade III variants: papillary, rhabdoid and anaplastic.
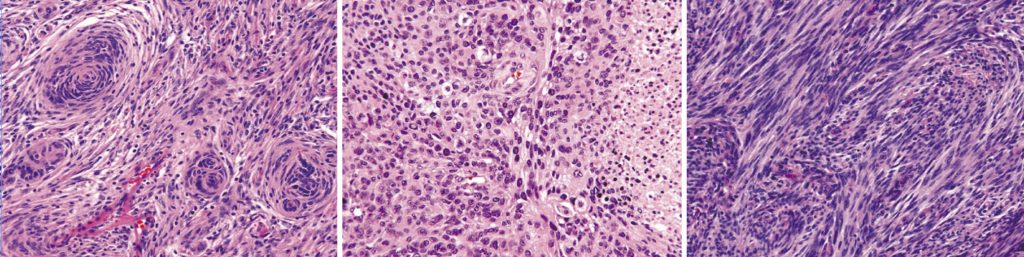
Peripheral nerve tumours
The previous entities schwannomas, WHO grade I (comprising schwannoma, plexiform schwannoma and cellular schwannoma variants), neurofibromas WHO grade I (comprising neurofibroma, atypical neurofibroma and plexiform neurofibroma variants), and perineurinomas WHO grade I have been retained. Melanotic schwannoma was a schwannoma variant, but has been recognised as a separate entity due to evidence of potential for malignant transformation in up to 10% of patients:20 to avoid mis-prognostication, these tumours have not received any grading. Hybrid nerve sheath tumours, WHO grade I, another new entity, are tumours that show a combined schwannoma, neurofibroma and perineurinoma pattern. Malignant peripheral nerve sheath tumours (MPNST) traditionally had grades II, III and IV and this has been changed to epithelioid MPNST and MPNST with perineurial differentiation instead.21 Epithelioid MPNSTs show a multilobulated appearance and strong S100 protein positivity. Loss of INI1 expression (67% cases)22 and rare BRAFV600E mutations raise the possibility of alternate, targeted treatment in these commonly excised, less chemosensitive tumours.23
Conclusion
The 2016 molecular WHO classification reflects the recent giant leaps in the understanding of the genetics of brain tumours and hopefully heralds an era of better defined clinical trials and personalised therapeutic options for brain tumour patients. It has not changed the surgical approach to most tumours in the CNS, which still hinges on either a diagnostic biopsy or maximal safe resection. The most important outcome of the new classification has been to highlight molecular targets for novel adjunctive treatments aiming to improve prognosis and survival. It has highlighted the fact that prognostic and survival figures previously estimated for CNS tumours might not be entirely accurate, and there needs to be a paradigm shift in the way we advise our patients about these tumours. However, the 2016 WHO classification has also highlighted the amount of work that is still necessary to achieve better genetically based classifications for many tumours such as meningiomas and ependymomas. Initiatives such as the 100,000 genomes project will further our understanding in this regard. It is vital that neuroscience clinicians stay abreast of these developments and work in a multidisciplinary team including neuropathologists, oncologists and neuroradiologists to conduct an informed, up to date practice.
References
- Virchow R. Die Krankhaften Geschwulste. Hirschwald 1863-67, Berlin. 1863;
- Bailey P CH. A classification of tumors of the glioma group on a histogenetic basis with a correlation study of prognosis. Lippincott, Philadelphia. 1926;
- Kernohan JW, Mabon RF, Svien HJ AA. A simplified classification of the gliomas. Symposium on a new simplified concept of gliomas. Mayo Clin Proc 71-75. 1949;
- Daumas-Duport C, Scheithauer B, O’Fallon J, Kelly P. Grading of astrocytomas. A simple and reproducible method. Cancer [Internet]. 1988 Nov 15;62(10):2152- 65. https://doi.org/10.1002/1097-0142(19881115)62:10<2152::AID-CNCR2820621015>3.0.CO;2-T
- Louis DN. The next step in brain tumor classification:Let us now praise famous;… or molecules? Acta Neuropathol [Internet]. 2012 Dec 15;124(6):761-2.
- Louis DN, Ohgaki H, Wiestler OD CW. World Health Organization Histological Classification of Tumours of the Central Nervous System. International Agency for Research on Cancer, France. 2016. https://doi.org/10.1007/s00401-012-1067-4
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK ED, Figarella-Branger D, Reifenberger G von DA. WHO classification and grading of tumours of the central nervous system. IARC Press Int Agency Res Cancer, Lyon. 2016;
- Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res [Internet]. 2013 Feb 15;19(4):764-72. https://doi.org/10.1158/1078-0432.CCR-12-3002
- Kurian KM, Haynes HR, Crosby C, Hopkins K, Williams M. IDH mutation analysis in gliomas as a diagnostic and prognostic biomarker. Br J Neurosurg [Internet]. 2013;27(August):1-4.
- Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol [Internet]. 2016 Apr 19;27(4):599-608. https://doi.org/10.1093/annonc/mdw013
- Kleinschmidt-DeMasters BK, Aisner DL, Birks DK, Foreman NK. Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol [Internet]. 2013 May;37(5):685-98.
- Perry A, Miller CR, Gujrati M, Scheithauer BW, Zambrano SC, Jost SC, et al. Malignant gliomas with primitive neuroectodermal tumor-like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol [Internet]. 2009 Jan;19(1):81-90. https://doi.org/10.1111/j.1750-3639.2008.00167.x
- Herrlinger U, Jones DTW, Glas M, Hattingen E, Gramatzki D, Stuplich M, et al. Gliomatosis cerebri: no evidence for a separate brain tumor entity. Acta Neuropathol [Internet]. 2016 Feb 22;131(2):309-19. https://doi.org/10.1007/s00401-015-1495-z
- Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J, Simon M, et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol [Internet]. 2010 Dec 19;120(6):707-18. https://doi.org/10.1007/s00401-010-0781-z
- Cancer Genome Atlas Research Network, Brat DJ, Verhaak RGW, Aldape KD, Yung WKA, Salama SR, et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med [Internet]. 2015 Jun 25;372(26):2481-98. https://doi.org/10.1056/NEJMoa1402121
- McNamara MG, Jiang H, Lim-Fat MJ, Sahebjam S, Kiehl T-R, Karamchandani J, et al. Treatment Outcomes in 1p19q Co-deleted/Partially Deleted Gliomas. Can J Neurol Sci / J Can des Sci Neurol [Internet]. 2017 May 9;44(03):288-94. https://doi.org/10.1017/cjn.2016.420
- Huse JT, Diamond EL, Wang L, Rosenblum MK. Mixed glioma with molecular features of composite oligodendroglioma and astrocytoma: a true “oligoastrocytoma”? Acta Neuropathol [Internet]. 2015 Jan 31;129(1):151-3. https://doi.org/10.1007/s00401-014-1359-y
- Ida CM, Rodriguez FJ, Burger PC, Caron AA, Jenkins SM S, GM, Aranguren DL, Lachance DH GC. Pleomorphic xanthoastrocytoma: natural history and long-term follow- up. Brain Pathol. 2015;(25):575- 586.
- Saraf S, McCarthy BJ, Villano JL. Update on meningiomas. Oncologist [Internet]. 2011;16(11):1604-13. https://doi.org/10.1634/theoncologist.2011-0193
- C arney JA. Psammomatous melanotic schwannoma. A distinctive, heritable tumor with special associations, including cardiac myxoma and the Cushing syndrome. Am J Surg Pathol [Internet]. 1990 Mar;14(3):206-22. https://doi.org/10.1097/00000478-199003000-00002
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol [Internet]. 2016 Jun 9;131(6):803-20. https://doi.org/10.1007/s00401-016-1545-1
- Jo VY, Fletcher CDM. Epithelioid malignant peripheral nerve sheath tumor: clinicopathologic analysis of 63 cases. Am J Surg Pathol [Internet]. 2015 May;39(5):673-82. https://doi.org/10.1097/PAS.0000000000000379
- Rekhi B, Kosemehmetoglu K, Tezel GG, Dervisoglu S. Clinicopathologic features and immunohistochemical spectrum of 11 cases of epithelioid malignant peripheral nerve sheath tumors, including INI1/SMARCB1 results and BRAF V600E analysis. APMIS [Internet]. 2017 Apr 27 https://doi.org/10.1111/apm.12702