




Abstract
We describe an unusual case of giant cell arteritis initially manifesting as insidiously progressive spastic quadriparesis, widespread muscle wasting and fasciculations in the absence of headache, followed by a complete left pupil-involving 3rd nerve palsy 10 months later. MRI and CSF analysis revealed evidence of intracranial involvement with established white matter lesions and intrathecal oligoclonal bands, respectively, whilst whole body FDG-PET demonstrated isolated uptake within the descending aorta. The temporal arteries were clinically and radiologically unremarkable but biopsy showed transmural inflammation and multinucleate giant cells. A rapid, complete and sustained improvement followed steroid therapy.
Case presentation
A 69-year-old male ex-smoker with well-managed hypertension presented with painless diplopia on right gaze 15 minutes after waking, followed by progressive left-sided ptosis which completed over four hours. Over the preceding 10 months he also reported increasing pain and weakness in both legs, with particular difficulty rising from his car seat and a significant limitation in walking distance. Four months previously similar symptoms had begun in the arms and the patient had noticed generalised loss of muscle bulk and that occasionally muscles would ‘twitch’. Appetite had become poor and body weight had fallen by 10kg, with sporadic episodes of nocturnal sweating. Bowel, bladder and cognitive functions were normal. Medications included Amlodipine 5mg daily and occasional Ibuprofen for osteoarthritis. Examination revealed a weakly reactive dilated left pupil, with globe exotropia and abduction, but normal visual acuity and fields, anterior and posterior chambers, and fundoscopy. The remaining cranial nerves were normal, apart from a brisk jaw jerk. Forward neck flexion was MRC 4/5 in the context of myalgia. However, there was generalised loss of muscle bulk, fasciculations in the limbs and upper trunk, and a spastic quadriparesis with MRC power 4/5 at elbow extension, hip flexion and knee flexion. Reflexes were globally brisk including finger jerks, Hoffman signs and crossed adductors, in addition to ankle clonus and extensor plantars bilaterally. Sensation to all modalities and co-ordination were normal. Cardiorespiratory and abdominal systems examined normally without rashes, ulcers or palpable masses.
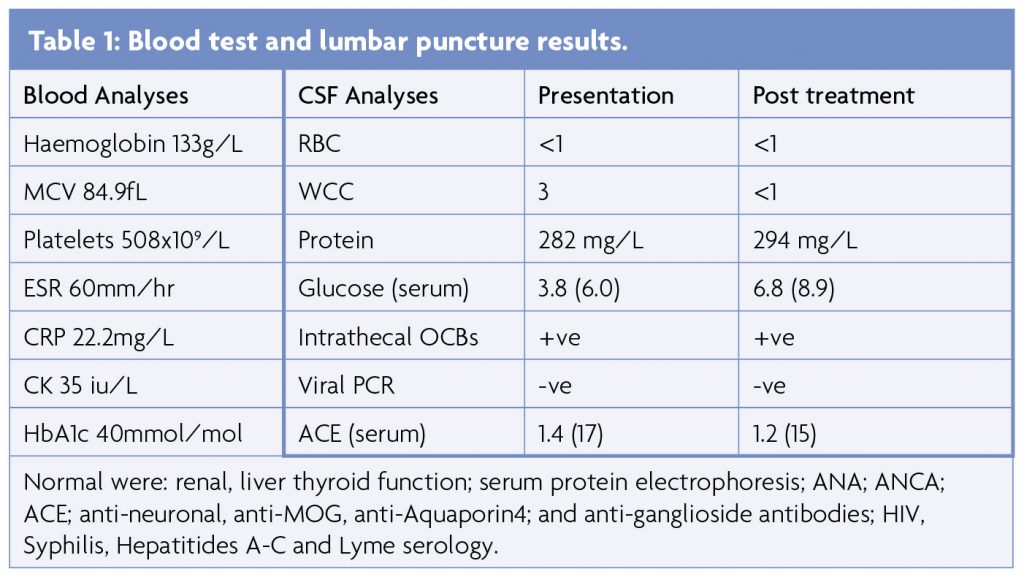
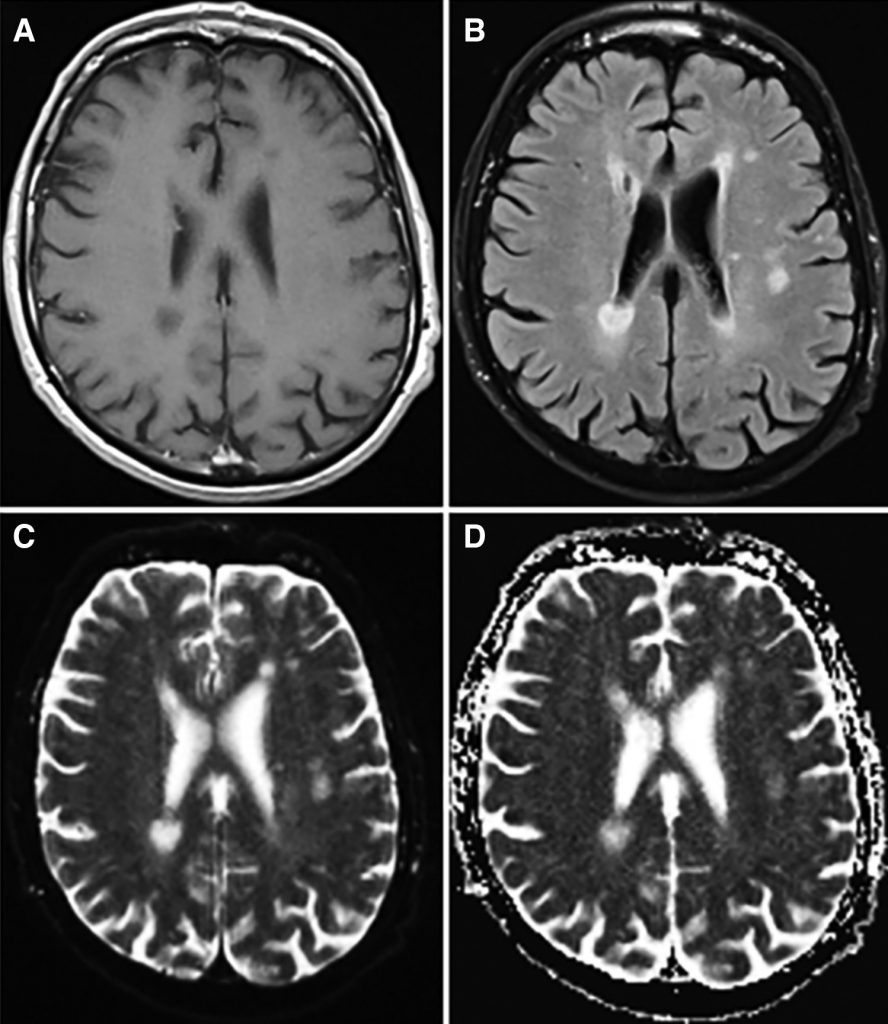
Blood results (Table 1) included mild normocytic anaemia, elevated platelets and raised ESR (peak 63mm/hr) and CRP (peak 70) in the absence of infection or autoantibodies. MRI brain and whole spine showed multiple areas of signal hyperintensity within the white matter and basal ganglia of both hemispheres without contrast enhancement or restricted diffusion (Figure 1), raising the possibility of inflammation or ischaemia. Initial CSF analysis was essentially unremarkable but an intrathecal pattern of oligoclonal bands synthesis was detected (Table 1). Neurophysiology confirmed widespread fasciculation potentials in the limbs, without polyphasic sharp waves or fibrillations and the tongue appeared normal. Mild neurogenic changes in the limbs were noted upon voluntary activation, with polyphasic waves and reduced recruitment. Normal motor and sensory conduction was otherwise seen with no evidence of myopathic discharges. CT with contrast of the chest, abdomen and pelvis was normal and whole body PET-CT was unremarkable, apart from a mild non-specific uptake along the left wall of the descending thoracic aorta considered at first to be a radiological artifact, given that it was not circumferential.
{kind=link}
The patient continued to deteriorate with fluctuating low grade pyrexia, myalgia and drenching night sweats. Nutritional supplements in addition to regular meals were only maintaining his already decreased body weight and limb weakness worsened such that he required a wheelchair for distances beyond a few meters. Interestingly, prior to steroid therapy his 3rd nerve palsy began to spontaneously improve over six to eight weeks.
Consideration of the differential diagnoses
The evolution of motor symptoms might have initially been suggestive of amyotrophic lateral sclerosis (ALS) or one of its mimic syndromes,1 although the progressive systemic symptoms and abrupt onset diplopia by the time of his presentation became inconsistent with these. The acute, unilateral, pupil-involving left oculomotor nerve palsy arising in the absence of contralateral ptosis, superior rectus paresis or concurrent hemiparesis pointed away from respective nuclear IIIrd, Benedikt’s fascicular or Weber’s fascicular midbrain syndromes and instead to a peripheral lesion outside the brainstem and proximal to its division within the superior orbital fissure. Accounting for pupil involvement, an external compressive cause would still have been a reasonable consideration over a microvascular phenomenon, although additional cranial nerve palsies and meningism were not present to suggest pathology within the cavernous sinus or basal meninges, where the imaging was unremarkable.
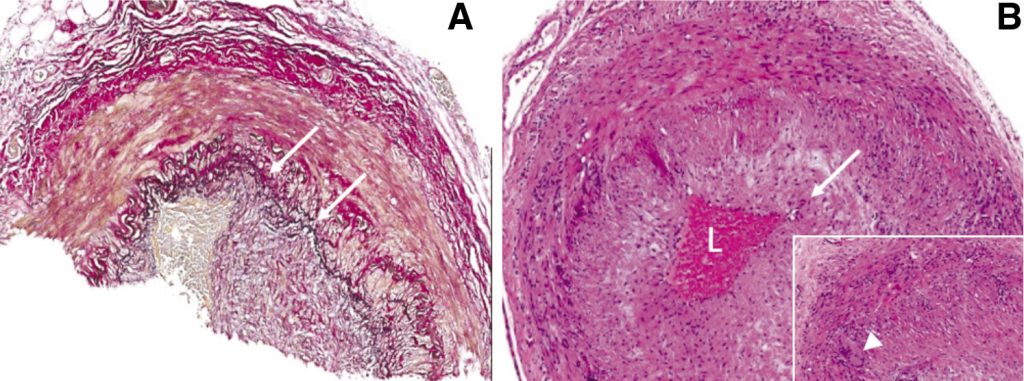
Clinical examination implicated bilateral pyramidal tract and spinal motor neuron involvement at multiple levels, as corroborated by central nervous system lesions on MRI and widespread neurogenic change on neurophysiology, respectively. Although no neuromyotonic discharges were seen on EMG, the predominantly proximal pattern of weakness with myalgia and systemic features prompted consideration of underlying endocrinological, infective, neoplastic, drug-induced, inflammatory muscle and neuromuscular junction disorders2 for which no clear evidence was found. However, given the equivocally abnormal FGD-PET findings, negative autoantibody screen (including anti-neuronal antibodies) and lack of organ involvement anticipated in other vasculitides, a large vessel vasculitis was suspected as the most plausible diagnosis to unify malaise, fever, night sweats, weight loss, myalgia, normocytic anaemia, raised ESR and CRP, clinically progressive CNS lesions and complete 3rd nerve palsy. Intra- and extracranial MR angiography were normal and the temporal arteries prominent but not pulseless or tender. Nevertheless, temporal artery biopsy demonstrated a mainly lymphocytic transmural inflammation, with occasional multinucleate giant cells (Figure 2). Of note, well formed non-necrotising granulomas were not present.
{kind=link}
A diagnosis of Giant Cell Arteritis (GCA) was made in accordance with The American College of Rheumatologists Classification (age >50, ESR >50mm/hr and abnormal artery biopsy).3 Intravenous Methylprednisolone 1g daily for three days was commenced immediately, followed by Prednisolone at 1mg/kg/day and Aspirin 75mg daily. Within 72 hours the patient reported cessation of myalgia and night sweats. Weight and walking distance improved over the next month, muscle twitching ceased and the ESR, CRP and haemoglobin normalised.
Patient outcome
After four weeks, Prednisolone was gradually tapered over eight months. Reducing the dose below 10mg/day on two occasions unmasked increasing leg discomfort, weakness, poor concentration, and swelling at the temporal biopsy site, which responded rapidly to dose restoration and a much more gradual reduction. Repeat neurophysiology at nine months was normal and serial MR neuraxis imaging has remained unchanged for 34 months.
Discussion
Giant cell arteritis (GCA) is a granulomatous autoimmune vasculitis typically affecting medium and large diameter vessels in proportionally more female patients over the age of 50 (peak incidence 70-79).3-6 Suspicion is classically raised by new onset headache or localised head pain in this demographic, and is the only heraldic symptom defined within currently accepted diagnostic criteria.3 Prompt initiation of glucocorticoids is obliged to prevent the feared complication of permanent visual loss secondary to anterior ischaemic optic neuropathy. Superficial temporal artery specimens are often used to confirm GCA pathologically, although up to 40% are unrevealing due to characteristically inhomogeneous segmental involvement with so-called ‘skip lesions’.7 Diagnosis can be made on clinical grounds but the condition’s alternative designations of “temporal arteritis” or “cranial arteritis” shouldn’t detract from the extracranial manifestations such as limb claudication. Indeed, headache may be conspicuously absent, as in this case, despite a positive tissue diagnosis. Furthermore, aching and stiffness in the pelvic and/or shoulder girdles distinctive for polymyalgia rheumatica (PMR) is seen in roughly 50% of patients with GCA at diagnosis and, conversely, around a fifth of patients with PMR develop GCA,8 with ‘constitutional’ symptoms of weight loss, fever and night sweats frequently encountered in both conditions.6
Histopathologically, the immune-mediated assault of arterial walls in GCA is believed to evolve from the outside in although which factors may initiate the whole cascade are unknown. Antigen-presenting dendritic cells within the adventitia enter a ‘mature’ activated state which enables recruitment of T cells via endothelium in the vasa vasorum followed by macrophages which may eventually coalesce into multinucleated giant cells.5,6,9 Pro-inflammatory cytokines including inter-leukin-6 (IL-6), IL-1 and interferon gamma sustain what becomes a transmural inflammatory process and are implicated in mediating the constitutional and myalgic symptoms in association with increased CRP, prolongation of the ESR, elevation of platelets and activated circulating monocytes.9,10 Arterial biopsies in PMR are rarely undertaken, but dendritic cell activation without a T cell response has been described, as have isolated clumps of mononuclear inflammatory cells surrounding capillaries proximal to the adventitia.11,12 For these reasons, large vessel imaging modalities such as axillary ultrasound, CT/MR angiography and FDG-PET are able to demonstrate increased arterial wall echogenicity, thickness or glucose uptake, respectively, supportive of a (perhaps subclinical) vasculitic process in patients with ‘PMR’ or in lieu of arterial biopsy,5,6 accepting that a less than overwhelming ‘artefactual’ or ‘equivocal’ anomaly might need to be interpreted in context, as here. Accordingly, it has been proposed that the diagnostic criteria for GCA as a syndrome be expanded to include affirmative imaging findings and the spectrum of cranial and extracranial features.6
This particular patient is unusual for several reasons. Extraocular paresis in GCA, albeit on the rarer side, is well recognised and almost always involves the oculomotor and/or abducens nerves. However, pupil involvement is not common.13,14 Despite one case previously incriminating extraocular muscle necrosis,15 occlusive arteritis affecting branches of the ophthalmic artery within the globe are considered responsible for paresis of eye movement14 and would certainly seem most plausible in our case. Indeed, diplopia is often reported in the days to weeks leading up to frank visual loss.14
The brain parenchymal lesions and the unmatched oligoclonal bands (OCBs) in the CSF are unusual. In a known hypertensive and ex-smoker it is possible that the subcortical lesions reflected accelerated microvascular damage. There was no evidence of abnormally restricted diffusion or contrast enhancement and, without a pre-morbid comparator, the changes are impossible to age. However, the progressive motor symptoms and signs in context of the diplopia and PMR suggest active central pathology related to GCA and might provide an explanation for the OCBs which are not unusual in CNS vasculitides. It is unclear how often positive OCBs are reported specifically in GCA even with cerebral involvement, although B-cell involvement is increasingly recognised.5,16 An incidental second diagnosis, such as late onset primary neuroinflammatory disease, would be unlikely given there has been sustained clinical improvement without radiological progression and we would prefer to apply Occam’s Razor here. An alternative diagnosis of neurosarcoid is worth consideration, however serum and CSF ACE were normal, there was no suggestion of systemic involvement on CT or PET imaging, there was no haemorrhagic component or enhancement of the CNS lesions or meninges and the ophthalmology assessment was not supportive of this diagnosis and as such we think a systemic vasculitic process remains most likely.
Finally, the widespread fasciculations are most intriguing. The authors are unable to find mention of these in conjunction with GCA but they clearly arose in the context of the myalgia and weakness, and evaporated quickly following steroid therapy. Neurophysiology additionally demonstrated minimal neurogenic changes and mildly reduced recruitment which is similar to that seen in partial compressive root lesions with proximally-generated fasciculations.17 We propose that the fasciculations were generated peripherally representing a multilevel radicular arteritis of the vasa nervora, albeit without evidence of significant axonal loss.
Learning Points
- Giant Cell Arteritis (GCA) can present with intra- or extracranial manifestations in the absence of headache.
- GCA is a vascular cause of pupil-involving complete 3rd nerve palsy.
- Large vessel imaging modalities, such as CT or MR angiography and FDG-PET, may support diagnosis of GCA.
- Positive oligoclonal bands may be seen with intracranial vessel involvement.
- Widespread fasciculations in GCA is highly unusual and may represent multilevel proximal radiculopathy.
References
- Turner MR, Talbot K. Mimics and chameleons in motor neurone disease. Practical Neurology. 2013;13:153-164. https://doi.org/10.1136/practneurol- 2013-000557
- Suresh E. & Wimalaratna, S. Proximal myopathy: diagnostic approach and initial management. Postgrad Med J 2103;89:470-477. https://doi.org/10.1136/postgradmedj-2013-131752
- Dasgupta B. Giant Cell Arteritis Guideline Development. Concise guidance: diagnosis and management of giant cell arteritis. Clin Med (Lond). 2010;10:381-386. https://doi.org/10.7861/clinmedicine. 10-4-381
- Gonzalez-Gay MA et al. Epidemiology of giant cell arteritis and polymyalgia rheumatica. Arthritis Rheum. 2009;61:1454-1461. https://doi.org/10.1002/art.24459
- Dejaco C, et al. Giant cell arteritis and polymyalgia rheumatica: current challenges and opportunties. Nat Rev Rheumato. 2017;13:578-592. https://doi.org/10.1038/nrrheum.2017.142
- Dejaco C, Duftner C, Buttgereit F, Matteson, EL & Dasgupta B. The spectrum of giant cell arteritis and polymyalgia rheumatica: revisiting the concept of the disease. Rheumatolo-gy (Oxford). 2017;56: 506-515. https://doi.org/10.1093/rheumatology/kew273
- Duhaut P et al. Biopsy proven and biopsy negative temporal arteritis: differences in clinical spectrum at the onset of the disease. Groupe de Recherche sur l’Arterite a Cellules Geantes. Ann Rheum Dis. 1999;58:335-341. https://doi.org/10.1136/ard.58.6.335
- Gonzalez-Gay MA, et al. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore) 2005;84:269-276. https://doi.org/10.1097/01md.0000180042.42156.d1
- Ghosh P, Borg FA & Dasgupta B. Current understanding and management of giant cell arteritis and polymyalgia rheumatica. Expert Rev Clin Immunol. 2010;6:913-928. https://doi.org/10.1586/eci.10.59
- van der Geest KSM, et al. Review: What Is the Current Evidence for Disease Subsets in Giant Cell Arteritis? Arthritis Rheumatol 2018;70:1366-1376 (2018). https://doi.org/10.1002/art.40520
- Chatelain D et al. Small-vessel vasculitis surrounding an uninflamed temporal artery: a new diagnostic criterion for polymyalgia rheumatica? Arthritis Rheum. 2008;58:2565-2573. https://doi.org/10.1002/art.23700
- Weyand CM, & Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med. 2003;349:160-169. https://doi.org/10.1056/NEJMra022694
- Warburton KL, Austen E, Gough A. & Wihl GE. An unusual cause of bilateral ophthal-moplegia. BMJ Case Rep (2010). https://doi.org/10.1136/bcr.09.2010.3353
- Fisher CM. Ocular palsy in temporal arteritis. I. Minn Med. 1959;42:1258-1268.
- Barricks ME, Traviesa DB, Glaser JS & Levy IS. Ophthalmoplegia in cranial arteritis. Brain. 1977;100:209- 221. https://doi.org/10.1093/brain/100.2.209
- Ciccia F, et al. Ectopic expression of CXCL13, BAFF, APRIL and LT-beta is associated with artery tertiary lymphoid organs in giant cell arteritis. Ann Rheum Dis. 2017;76:235-243. https://doi.org/10.1136/annrheumdis-2016-209217
- de Carvalho, M & Swash, M. Origin of fasciculations in root lesions. Clin Neurophysiol. 2016;127: 870-873 (2016). https://doi.org/10.1016/j.clinph.2015.02.004