


Summary
The visual and vestibular systems together mediate the reflex and perceptual functions required for efficient postural balance and spatial orientation in the light or the dark. Following an acute unilateral vestibular insult, there is a left-right imbalance in the vestibular input leading to erroneous brainstem vestibular signalling.
This asymmetrical vestibular signalling manifests in several ways, e.g. activation of reflex eye movements (vestibular nystagmus) and sensations of vertigo. The net result is a functional impairment in the capacity to locomote and spatially orientate; a high risk situation in a natural environment. The central nervous system can however promote a rapid functional recovery by shifting the brain’s relative reliance from vestibular toward visual cues for these functions, i.e. the brain becomes more ‘visually dependent’ for the sensory monitoring of locomotion and spatial orientation. Given time and the appropriate conditions (e.g. adequate physical activity, avoidance of vestibular sedatives), the fidelity of the brainstem vestibular signal is restored as the brainstem process involved in ‘vestibular compensation’ removes the left-right imbalance in the vestibular brainstem signal. In patients who make a full symptomatic recovery consequent upon adequate ‘vestibular compensation’, the reliance on visual signals for locomotion and navigation reduces toward normal levels in tandem with the process of brainstem vestibular compensation. In contrast, some chronically symptomatic patients show a maladaptive persistence of this heightened visual dependency and this is manifest in visually-induced dizziness (“visual vertigo”). Visually-induced dizziness is a major problem in the clinic, with symptoms occurring in visually-busy environments such as shopping malls or supermarkets. We discuss the physiological mechanisms underlying visual-vestibular interaction, how this interaction may be disturbed leading to visually-induced dizziness and finally how understanding the physiological mechanism helps in the development of therapy for these patients.
Introduction
The vestibular system, which provides a signal of head motion to the brain, mediates functions of gaze and postural stabilisation via vestibular-ocular (VOR) and vestibular-spinal reflexes. The vestibular system is also key in generating sensations of self-motion and spatial orientation required for navigation in the environment. The vestibular system influences these reflex and perceptual functions in partnership with other sensory systems, particularly vision. For example, vision calibrates the accuracy of the VOR and, via optic flow and motion parallax generated during self-motion, contributes to our sense of self-motion (or stasis).
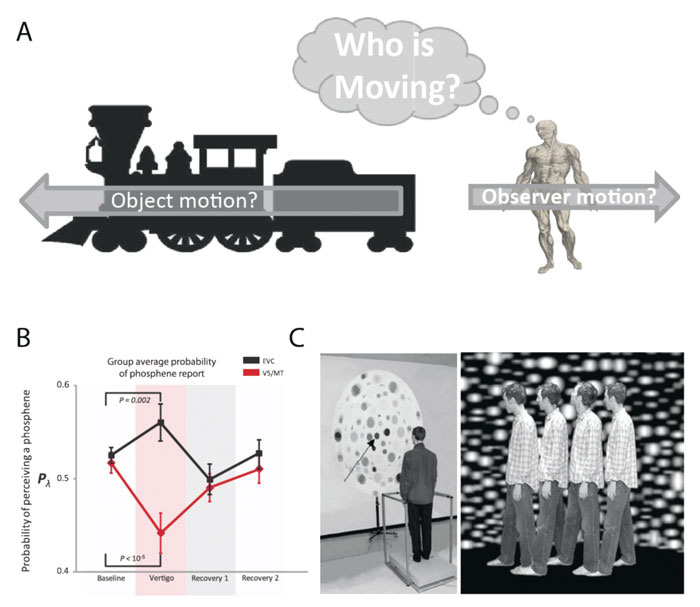
Occasionally, visual-vestibular interaction can mislead, e.g. the compelling but false sensation of self-motion experienced when looking out of the window of a stationary train as an adjacent train moves past us (‘the ‘train illusion’, Figure 1A). This illusion demonstrates the difficulty the brain has in trying to resolve the complex and ambiguous role of the visual system in signalling both self- and object (environmental) motion.
The occasional failure of the normal brain to accurately estimate measures of self-motion is key to understanding how many patients’ symptoms relate to an abnormal visuo-vestibular interaction. Indeed patients with vestibular disorders commonly report a modulation of their dizziness by visual stimuli. In acute vertigo, where typically there are abnormal signs such as a vestibular nystagmus,1 patients often close their eyes to avoid the distressing illusion of seeing the world spinning. In contrast, in chronic dizziness where there are usually no abnormal signs (an apparent uncoupling of symptoms from signs), patients complain of dizziness in the face of relatively trivial motion in the environment (e.g. crowds in a shopping mall). Since such visual stimuli are ubiquitous in the modern world, visually-induced dizziness (so-called ‘visual vertigo’) may be crippling for patients’ social, occupational and mental well-being. In this overview we explore the basic mechanisms underlying the intimate relationship between visual motion and dizziness, and the relevance of this visual-vestibular interaction for patients’ symptoms and their management.
Role of the vestibular system in health
As we navigate through the environment, our visual system is faced with two challenges: (1) the maintenance of a stable and clear image of the world during head movements; (2) the accurate ascribing of visual motion as being due to either self-motion or environmental motion – put simply the brain asks the question: am I moving? or is the object/world moving? To overcome these problems the central nervous system combines visual and vestibular inputs.
Maintaining a clear and stable vision is enabled by a natural ‘steady-cam’ mechanism called the vestibular-ocular reflex (VOR).2 The VOR involves a 3-neurone brainstem reflex that begins with the detection of head acceleration by the peripheral labyrinth. This head motion signal is conveyed by primary vestibular afferents to the vestibular nuclei neurones in the brainstem which in turn project to ocular motor neurones. The VOR thus keeps the eyes steady and ‘locked on’ to the visual target of interest despite head motion. This mechanism thus maintains visual acuity and a stable visual world by reducing slippage of the visual image across the retina. This ‘retinal slip’, when it does occur, may provoke the unpleasant sensation of oscillopsia. In general the degree of oscillopsia is coupled to the amount of retinal slip, particularly in the acute state. Retinal slip and oscillopsia symptoms are not however inevitably linked but can be uncoupled in the chronic adapted state.3-5 The capacity for the brain to render a physical retinal slip unnoticeable is an important concept since it leads to the finding that ocular motor (reflex) parameters of vestibular function (i.e. VOR) relate poorly to perceptual aspects of vestibular function (i.e. dizziness) in the chronic state.4 Indeed a relatively common but extreme example of such perceptuo-reflex uncoupling is that seen in idiopathic congenital nystagmus where a vigorous nystagmus is not associated with symptoms.5
The visuo-vestibular interaction
One mechanism proposed to solve the motion ambiguity problem is that of a reciprocal visual-vestibular inhibition (Figure 2). Specifically, if the vestibular system signals ‘no motion’, then this impedes a visual motion signal from indicating self-motion. Conversely, when there is no vestibular signal, visual input can provoke a sensation of self-motion but only if the visual stimulus occupies a sufficiently large visual area. Cognitive influences are also important since illusory self-motion is more likely to occur if there is a high probability of self-motion, e.g. sitting on a train is a situation where motion is likely, whereas sitting on the sofa has a low probability of motion.
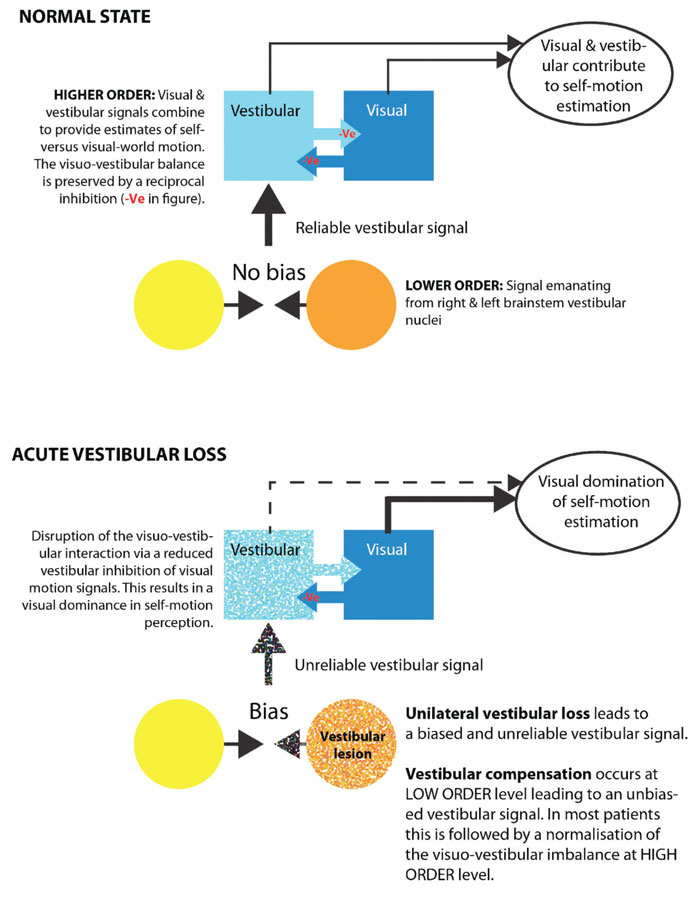
In the normal state visual and vestibular signal combine to provide estimates of self-versus visual-world motion. Following acute vestibular loss, the imbalance of inputs from the vestibular system results in an unreliable vestibular signal. Therefore during this period the brain relies more heavily on visual information.
The psychophysical evidence for a visuo-vestibular reciprocal inhibition is supported by behavioural data showing that during self-motion, the threshold for visually detecting object-motion is elevated.6,7 Similarly visual-vestibular reciprocal inhibition is invoked to explain the observation that vestibular stimulation can disrupt performance on visualisation and mental rotation tasks.8 Note that although vision is the critical sensory modality for the normal calibration of vestibular signals, the vestibular system can utilise non-visual sensory signals as evidenced by the ability of congenitally blind individuals to orientate themselves using only vestibular cues of motion.9
The neural basis for the higher order (perceptual) brain response during visuo-vestibular interaction has been explored using functional imaging,10 lesion mapping11 and brain stimulation.12 Unlike the motor or somatosensory systems, there is no primary vestibular cortex, rather vestibular signals are widely conveyed to the cerebral cortex.13,14 Conversely vestibular sensitive cortical neurones invariably display reactivity to other sensory inputs such as proprioception or visual motion, i.e. vestibular sensitive neurones are truly multi-modal sensory neurones.13,15
Vestibular stimulation e.g. via bithermal caloric irrigation or galvanic stimulation of the vestibular nerve, is associated with increased neuro-imaging signal in a network of brain regions primarily in the Sylvian fissure, insula, retroinsular cortex, fronto-parietal operculum, superior temporal gyrus and cingulate cortex.16-19 Conversely, signal reduction is observed in visual cortex (the neuro-imaging correlate of visual-vestibular reciprocal inhibition). In contrast optokinetic visual stimulation inducing vection engenders an opposite pattern, i.e. reduced signal in somatosensory and parietal (‘vestibular’) areas versus increased signal in visual cortex.20-22
Since neuroimaging is a correlational technique we recently utilised transcranial magnetic stimulation (TMS) to probe visual cortical excitability during vestibular activation (see Figure 1B and reference 12). We posed two main questions: first, is there a true change in visual cortical excitability during vestibular activation; second, is the visual cortical response uniform or is there a differential response between early visual cortex (includes V1/2) and visual motion cortex (area V5/MT). This latter aspect was prompted by the lack of clarity in the literature with some suggesting a uniform visual cortical involvement,16,23 versus those proposing a selective involvement17,24 of visual motion areas. In this experiment, we probed the excitability of visual motion area V5/MT and separately early visual cortex (EVC), i.e. areas V1 and V2, using TMS, during vestibular activation (obtained by caloric stimulation). TMS can be used to probe visual cortical excitability by measuring the relative ease with which one can evoke a phosphene25 (a perceived flash of light elicited by visual cortex electrical or magnetic stimulation26). We found that vestibular stimulation was associated with decreased V5/MT excitability versus increased excitability of early visual cortex (see Figure 1B). Thus, strong stimulation of the vestibular system may reduce sensitivity of visual motion detection areas, but crucially leaves early visual cortex functionally intact (thus not interfering with visual discriminative functioning). This finding provides a possible neurophysiological correlate for the putative reciprocal inhibition between vestibular and visual cortical networks.
Functional imaging has also been used to compare the brain activation of healthy controls during vestibular stimulation to activity in patients following a vestibular lesion. Patients with vestibular neuritis were examined using positron-emission tomography during the acute stage and again three months later. Increases in regional cerebral glucose metabolism (rCGM) were found in the acute stage relative to the recovery period in multisensory vestibular cortical areas, whereas reduced rCGM was reported in visual and somatosensory areas.27 These acute stage activation patterns largely mirror those described during vestibular stimulation in healthy volunteers.28
The relationship between brain structure and vestibular function has also been investigated using neuroimaging. In a follow-up study, patients with VN were tested at least six months after disease onset. Increased grey matter density was reported in medial vestibular nuclei and right gracile nucleus, and increased white matter in the pontine commissural fibres, whereas reduced grey matter was found in left hippocampus and right superior temporal gyrus. Patients who reported residual canal paresis also demonstrated increased grey matter in MT/V5 regions.29 A recent study investigated associations between variability in grey matter changes and clinical outcomes in patients with unilateral vestibular failure.29 Grey matter density in the patients was compared to age-matched controls and revealed signal increases in medial vestibular nuclei and the right gracile nucleus grey matter, and reductions in left posterior hippocampus and the right superior temporal gyrus. Patients who demonstrated a significant residual canal paresis also showed increased grey matter density bilaterally in visual-motion sensitive areas in middle temporal cortex (MT/V5), an area that also receives vestibular input.30 This may reflect an attempt to compensate for (vestibular) motion sensitive deficits experienced by patients with significant vestibular deficits after vestibular neuritis.31 Outcomes as measured by the clinical vestibular score and subjective vestibular disability score were positively correlated with grey matter density in insular, retroinsular and MT and STG regions. These studies indicate that both brain functional and structural changes may take place during central vestibular compensation. Similarly, functional31-33 and imaging34 changes in visual mechanisms develop in patients with bilateral vestibular failure (e.g. secondary to gentamicin or idiopathic;35) probably underpinning adaptation to the oscillopsia experienced by bilateral vestibular patients.
Clinical relevance – ‘visually-induced dizziness’
The visuo-vestibular interaction is of particular clinical relevance to patients suffering from visually-induced dizziness36-38 previously known as visual vertigo, ‘visuo-vestibular mismatch’39 or ‘space and motion discomfort’.40 Patients with visually-induced dizziness report dizziness, unsteadiness and disorientation in visually disorienting surroundings but typically not classical rotational vertigo. The distinguishing characteristic of these patients compared to other dizziness patients is their tendency to be over reliant upon vision for postural control and balance, a situation termed ‘visual dependency’.37 Visually-induced dizziness appears to be the end result of repeated exposure to dizziness developing in diagnoses as disparate as vestibular migraine (see consensus statement on diagnosis of vestibular migraine41) BPPV or post-vestibular neuritis.
Visually-induced dizziness occurring post-vestibular neuritis should be distinguished from symptoms related to ‘poor compensation’ of the acquired unilateral peripheral vestibular deficit due to the vestibular neuritis. Poor compensation from a post-vestibular neuritis vestibular deficit can arise due to intermittent vertigo from BPPV, physical inactivity and/or excessive vestibular sedative therapy. A clear identification of the triggers (visual and non-visual) of symptoms post-vestibular neuritis is important since this determines the therapeutic intervention to alleviate these symptoms, e.g. stopping vestibular sedatives, initiating anti-migraine drugs, positional manoeuvres for BPPV, and vestibular physiotherapy, be it Cawthorne Cooksey exercises or optokinetic stimulation for visually-induced dizziness.
The profile of a typical patient with visually-induced dizziness would be a previously asymptomatic individual who suffered an acute vestibular insult such as vestibular neuritis (VN). We also see visually-induced dizziness in any patient with chronic recurrent dizziness, e.g. vestibular migraine (vestibular migraine is now an accepted International Headache Society diagnosis)41 or benign paroxysmal positional vertigo.
In a patient with post-vestibular neuritis visually-induced dizziness, typically as the patient’s overt continuous spinning vertigo abates another form of dizziness gradually increases whereby dizziness occurs in visually-busy situations such as supermarkets or shopping malls (leading to the false conclusion that the patient has a primary agoraphobia). Sometimes the patient does not discriminate between the acute attack of vertigo and subsequent chronic dizziness, and reports an acute onset with failure to improve over subsequent months. These patients may fail to report visual motion exacerbation of symptoms so it is important to always ask about ‘supermarkets’, ‘shopping malls’, ‘moving trains’, ‘action films’ or ‘video games’ as triggers of dizziness.
In Figure 2 we outline a hypothetical schema of the brain mechanisms underlying visual-vestibular interaction and visually-induced dizziness. An acute vestibular lesion results in an unreliable vestibular signal leading to impaired visual and postural stability. This unreliable vestibular signal is partially corrected for by the brain shifting its reliance from vestibular to visual motion information (‘visual dependency’). This acute visual dependence usually remits once the tonic vestibular imbalance resolves (via a process of rapid brainstem plasticity). Occasionally, this visual dependence persists despite an adequate rebalancing of the vestibular signal leading to a maladaptive state of visually-induced dizziness. Why some patients go on to develop long term visual dependency and visually-induced dizziness is not completely understood however investigation of the mechanisms of brain plasticity responsible for these symptoms are on-going.37,42
Whatever the neurobiological mechanisms underlying visually-induced dizziness and visual dependency, important aggravators of visually-induced dizziness include psychological symptoms,43 and migraine. How these aspects affect brain plasticity involved in vestibular compensation for an acute or recurring vestibular insult is unclear.44,45 Our recent data in individuals adapted to chronic vestibular stimulation suggest however that vestibular cerebellar plasticity plays a critical role in modifying the perceptual response to vestibular stimulation.46
Rehabilitation
Rehabilitation works by inducing plastic change in the brain. This plasticity changes the functional characteristics of the brain enabling normal function in the face of a prior insult (e.g. peripheral vestibular loss). Thus initial advice given to patients following an acute vestibular problem is a form of rehabilitation. Vestibular patients are recommended to continue with normal daily activities to ensure they are exposed to visual and vestibular challenges since such sensory stimulation drives the adaptive change required for symptomatic recovery. An important clinical point is that chronic treatment (>3 days) with vestibular sedatives is inimical to the recovery of vestibular patients, presumably by impairing the normal mechanisms of vestibular compensation. In chronic patients rehabilitation regimes should be adjusted to address the nature of the vertigo experienced. There is evidence from experiments in both healthy volunteers and chronically dizzy patients that rehabilitation therapy using optokinetic stimuli is an effective treatment.47,48 The first RCT study in this field examined the efficacy of simulator-based therapy in addition to customised treatment in a group of chronic unilateral vestibular patients who had responded poorly to conventional vestibular rehabilitation. Interventions included a planetarium and optokinetic disc stimuli (See Figure 2C) in order to examine whether visual dependence could be modulated in the patient group. The authors found significant improvements in visual vertigo symptom scores only in the group receiving the additional optokinetic simulator therapy.
The science underlying the effects of such physiotherapy is scarce. Recent laboratory data using TMS suggests that adapting to random visual motion promotes an acute increase of area V5/MT excitability, thus demonstrating the impact of visual motion stimulation upon visual cortical excitability. The use of a random motion stimulus contrasts with the OKN-type stimuli used in vestibular therapy sessions, however random motion stimulation may be ecologically relevant given patients’ symptoms of visual vertigo in situations where the visual motion is ‘Brownian’ (e.g. shopping malls). Whatever the nature of the visual motion stimulus, the observed modulation of cortical excitability may plausibly mediate the therapeutic effect of current clinical rehabilitation protocols that have been developed empirically.51
These studies suggest that rehabilitation training and exposure to visual stimuli may improve the symptoms of chronically dizzy patients by addressing the imbalance in their visuo-vestibular interaction and visual dependency. It is recommended that dizzy patients also pursue behaviours that challenge their visual dependence in addition to any formal rehabilitation they take part in. This is important as ‘real-world’ phenomena can never be fully replicated in the lab. Particularly helpful sports include those requiring VOR-smooth pursuit integration, e.g. ball sports such as tennis.
Clinical overview
An understanding of visuo-vestibular interaction and the underlying brain mechanisms is key in understanding patients’ superficially bizarre complaints (‘I feel dizzy when faced with shopping mall crowds or walking down supermarket aisles’) and secondly in developing effective treatment for visual vertigo. One potentially problematic group are vestibular migraineurs who frequently also complain of visually-induced dizziness. When treating such patients it is imperative to follow a step-wise approach. The first step is to treat the migraine with effective prophylaxis. We find that standard anti-migrainous drugs work well with propanolol being our first line (second line according to patient profile; including amitriptyline, topiramate, sodium valproate or pizotifen). Often simply treating the vestibular migraine with pharmacotherapy improves the visual symptoms as well. If visually-induced dizziness persists despite good migraine control, we then initiate OKN therapy. If OKN is provided to active migraineurs then symptoms can be aggravated, hence the importance of the first step (in treating the migraine). Once commenced on effective anti-migraine prophylaxis OKN therapy can be provided if symptoms of visually-induced dizziness persist. Indeed migraineurs show the greatest improvement in response to OKN therapy compared to patients with other chronic peripheral vestibular symptoms.48 Note however that the clinician should be alert to patients with psychological symptoms who also avoid visually busy environments but for different reasons, e.g. agoraphobia50. Equally many vestibular patients suffer from psychological symptoms as a result of their vestibular symptoms. In cases of doubt, a liaison psychiatric opinion should be sought. Needless to say, some patients require a two-pronged vestibular and psychological therapy approach. As always in medicine, a diagnosis and appropriate treatment have to be decided on multiple aspects of the clinical history and investigations.
Conclusion
An understanding of the brain mechanisms mediating visual and vestibular interaction has been little studied however multi-modal research involving neuroimaging, lesion mapping and more recently with TMS has enabled a mechanistic explanation for patients’ symptoms and the logical development of their treatment. There are many unanswered pertaining to the modulators of visual-vestibular interaction, such as migraine, anxiety and co-existing medical and neurological disorders.
References
- Seemungal BM. Neuro-otological emergencies. Current opinion in neurology 2007;20:32-9.
- Waespe W & Henn V. Neuronal activity in the vestibular nuclei of the alert monkey during vestibular and optokinetic stimulation. Exp Brain Res 1977;27:523-38.
- Bronstein AM. Vision and vertigo. Journal of neurology 2004;251:381-7.
- Palla A, Straumann D. & Bronstein AM. Vestibular neuritis: vertigo and the high-acceleration vestibulo-ocular reflex. J Neurol 2008:2551479-82.
- Dell’Osso LF & Leigh RJ. Foveation period stability and oscillopsia suppression in congenital nystagmus: an hypothesis. Neuro-ophthalmology 1992;12169-83.
- Probst T, Brandt T & Degner D. Object-motion detection affected by concurrent self-motion perception: psychophysics of a new phenomenon. Behav Brain Res 1986;22:1–11.
- Probst T, Straube A & Bles W. Differential effects of ambivalent visual-vestibular-somatosensory stimulation on the perception of self-motion. Behav Brain Res 1985;16;71-9.
- Mast FW, Merfeld DM. & Kosslyn SM. Visual mental imagery during caloric vestibular stimulation. Neuropsychologia 2006;44;101-9.
- Seemungal BM, Glasauer S, Gresty MA & Bronstein AM. Vestibular perception and navigation in the congenitally blind. Journal of neurophysiology 2007;97:4341-56.
- Kleinschmidt A, et al. Neural correlates of visual-motion perception as object-or self-motion. Neuroimage 2002;16:873-82.
- Kaski D, Bronstein AM, Malhotra P, Nigmatullina Y & Seemungal BM. Humans use an internal clock for estimating their position in space. in Society for Neuroscience, New Orleans, LA, 828.11, (2012).
- Seemungal BM, Guzman-Lopez J, Arshad Q, Schultz SR, Walsh V, Yousif N. Vestibular Activation Differentially Modulates Human Early Visual Cortex and V5/MT Excitability and Response Entropy. Cereb Cortex 2013;12-9.
- Grüsser OJ, Pause M & Schreiter U. Localization and responses of neurones in the parieto-insular vestibular cortex of awake monkeys (Macaca fascicularis). The Journal of physiology 1990;430:537-57.
- Guldin WO & Grüsser OJ. Is there a vestibular cortex? Trends in neurosciences 1998;21:254-9.
- Grüsser OJ, Pause M & Schreiter U. Vestibular neurones in the parieto-insular cortex of monkeys (Macaca fascicularis): visual and neck receptor responses. The Journal of physiology 1990;430:559-83.
- Wenzel R, et al. Deactivation of human visual cortex during involuntary ocular oscillations. Brain 1996;119:101-10.
- Bense S, Stephan T, Yousry TA, Brandt T & Dieterich M. Multisensory cortical signal increases and decreases during vestibular galvanic stimulation (fMRI). J Neurophysiol 2001;85:886-99.
- Naito Y, et al. Cortical correlates of vestibulo‐ocular reflex modulation: a PET study. Brain 2003;126:1562-78.
- Lopez C, Blanke O & Mast FW. The human vestibular cortex revealed by coordinate-based activation likelihood estimation meta-analysis. Neuroscience (2012).
- Brandt T, Bartenstein P, Janek A & Dieterich M. Reciprocal inhibitory visual-vestibular interaction. Visual motion stimulation deactivates the parieto-insular vestibular cortex. Brain 1998;121:1749-58.
- Bense S, et al. Direction‐dependent visual cortex activation during horizontal optokinetic stimulation (fMRI study). Hum Brain Mapp 2006;27:296-305.
- Kikuchi M, et al. Cortical activation during optokinetic stimulation-an fMRI study. Acta Otolaryngol 2009;129:440-3.
- Bottini G, et al. Cerebral representations for egocentric space Functional–anatomical evidence from caloric vestibular stimulation and neck vibration. Brain 2001;124:1182-96.
- Fasold O, et al. Human vestibular cortex as identified with caloric stimulation in functional magnetic resonance imaging. Neuroimage 2002;17:1384-93.
- Boroojerdi B, Prager A, Muellbacher W. & Cohen LG. Reduction of human visual cortex excitability using 1-Hz transcranial magnetic stimulation. Neurology 2000;54:1529-31.
- Brindley GS & Lewin WS. The sensations produced by electrical stimulation of the visual cortex. The Journal of physiology 1968;196:479-93.
- Bense S, et al. Preserved visual–vestibular interaction in patients with bilateral vestibular failure. Neurology 2004;63:122-8.
- Smith AT, Wall MB & Thilo KV. Vestibular inputs to human motion-sensitive visual cortex. Cerebral Cortex 2012;22:1068-77.
- Helmchen C, et al. Structural changes in the human brain following vestibular neuritis indicate central vestibular compensation. Annals of the New York Academy of Sciences 2009;1164, 104-15.
- Zu Eulenburg P, Stoeter P & Dieterich M. Voxel‐based morphometry depicts central compensation after vestibular neuritis. Annals of neurology 2010;68:241-9.
- Cousins S, et al. Vestibular Perception following Acute Unilateral Vestibular Lesions. PLOS ONE 2013;8:e61862.
- Bronstein AM, Morland AB, Ruddock KH & Gresty MA. Recovery from bilateral vestibular failure: implications for visual and cervico-ocular function. Acta Oto-Laryngologica 1995;115:405-7.
- Kalla R, et al. Adaptive motion processing in bilateral vestibular failure. Journal of Neurology, Neurosurgery & Psychiatry 2011;82;1212-16.
- Dieterich M, Bauermann T, Best C, Stoeter P & Schlindwein P. Evidence for cortical visual substitution of chronic bilateral vestibular failure (an fMRI study). Brain 2007;130:2108-16.
- Rinne T, Bronstein AM, Rudge P, Gresty MA & Luxon LM. Bilateral loss of vestibular function. Acta Oto-Laryngologica 1995;115:247-50.
- Bronstein AM. Visual vertigo syndrome: clinical and posturography findings. Journal of Neurology, Neurosurgery & Psychiatry 1995;59:472-6.
- Guerraz M, et al. Visual vertigo: symptom assessment, spatial orientation and postural control. Brain 2001;124:1646-56.
- Bisdorff A, Von Brevern M, Lempert T & Newman-Toker DE. Classification of vestibular symptoms: towards an international classification of vestibular disorders. Journal of Vestibular Research 2009;19:1-13.
- Longridge NS, Mallinson AI & Denton A. Visual vestibular mismatch in patients treated with intratympanic gentamicin for Meniere’s disease. Journal of otolaryngology 2002;31:5-8.
- Furman JM & Jacob RG. A clinical taxonomy of dizziness and anxiety in the otoneurological setting. Journal of anxiety disorders 2001;15:9-26.
- Lempert T, Olesen J, Furman J, Waterston J, Seemungal B, Carey J, Bisdorff A, Versino M, Evers S, Newman-Toker D. Vestibular migraine: Diagnostic criteria. Journal of Vestibular Research 2012;22:167-72.
- Keshner E, Streepey J, Dhaher Y & Hain T. Pairing virtual reality with dynamic posturography serves to differentiate between patients experiencing visual vertigo. J Neuroeng Rehabil 2007;4:24.
- Staab JP. Chronic dizziness: the interface between psychiatry and neuro-otology. Current opinion in neurology 2006;19:41-8.
- Drummond PD. Triggers of motion sickness in migraine sufferers. Headache 2005;45:653-6.
- Agarwal K, et al. Visual dependence and BPPV. Journal of neurology 2012;259:1117-24.
- Nigmatullina Y, Hellyer PJ, Nachev P, Sharp DJ & Seemungal BM. Attenuation of self-motion perception relates to reduced cortical connectivity. in Society for Neuroscience, New Orleans, LA; 828.03, (2012).
- Pavlou M. The Use of Optokinetic Stimulation in Vestibular Rehabilitation. Journal of Neurologic Physical Therapy 2010;34:105.
- Pavlou M, Bronstein AM & Davies RA. Randomized Trial of Supervised Versus Unsupervised Optokinetic Exercise in Persons With Peripheral Vestibular Disorders. Neurorehabilitation and Neural Repair (2012).
- Guzman-Lopez J, Silvanto J & Seemungal BM. Visual motion adaptation increases the susceptibility of area V5/MT to phosphene induction by transcranial magnetic stimulation. Clinical Neurophysiology 2011;122:1951-5.
- Staab JP. Chronic Subjective Dizziness. CONTINUUM: Lifelong Learning in Neurology 2012;18:1118-41.
- Pavlou M, Lingeswaran A, Davies RA, Gresty MA & Bronstein AM. Simulator based rehabilitation in refractory dizziness. Journal of neurology 2004;251:983-95.