

Summary
- The symptoms of Alzheimer’s disease are preceded by a long period of gradual accrual of pathological change. Familial Alzheimer’s disease, due to autosomal dominantly inherited mutations in the Presenilin 1 (PSEN1), PSEN2 and amyloid precursor protein (APP) genes, has provided important insights into the trajectory of imaging changes during this preclinical phase of disease.
- Studies of presymptomatic mutation carriers using a variety of imaging modalities have demonstrated that amyloid deposition, hypometabolism, atrophy and altered structural and functional connectivity are evident at different points in the years before expected age at symptom onset.
- Longitudinal imaging will be particularly important for further investigating the variable vulnerability of different brain regions to amyloid accumulation and the dynamics of change in different biomarkers through the presymptomatic phase.
- Presymptomatic prevention trials for familial Alzheimer’s disease using amyloid-modifying therapies are now being launched, in which imaging will play a central role.
It is now well established that the pathophyiological process of Alzheimer’s disease (AD) begins many years, even decades, before symptoms develop and there is currently great interest in defining and characterising the preclinical stages of the disease.1 This interest has been driven in part by the disappointing results of trials of amyloid-modifying therapies in patients with so-called “mild to moderate” AD, with at least one trial (of Solanezumab) showing in a post-hoc analysis that milder subjects appeared to do better than more affected individuals.2,3 The suggestion being that treatment in the majority of trials may have been “too little, too late” with the implication that treating earlier in the disease would have had greater chances of slowing progression. The observation from some previous trials, of apparent treatment-related reductions in amyloid-beta (Aβ) burden at autopsy or on amyloid imaging, have encouraged the view that disease-modification may be possible, but perhaps with only limited benefit if downstream neurodegeneration is already well established. 20-40% of cognitively normal older individuals show evidence of Aβ accumulation in both positron emission tomography (PET) imaging and cerebrospinal fluid studies and a similar proportion of healthy elderly people have been found to have AD pathology at post-mortem1 – the percentage who are “amyloid-positive” is very age-related and is also higher in those who carry an ApoE4 allele. The proportion of these individuals who will or would have gone on to develop AD dementia is currently unknown, so research recommendations have proposed the term ‘asymptomatic at risk state for AD’ to refer to this particular preclinical stage.4 The other proposed preclinical stage of AD is ‘presymptomatic AD, a term reserved for individuals with autosomal dominantly inherited mutations in the Presenilin 1 (PSEN1), PSEN2 and amyloid precursor protein (APP) genes who will inevitably develop symptoms of familial AD (FAD). In this review, we describe the insights into presymptomatic AD that have been gained through imaging studies of FAD mutation carriers and discuss some of the uncertainties and future challenges that remain as we enter an era of prevention trials for AD.
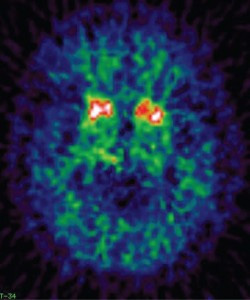
Understanding the timing and temporal order by which different imaging biomarkers become abnormal in presymptomatic AD is a fundamental issue for the field. The longitudinal study of healthy FAD mutation carriers with multi-modal imaging provides a unique opportunity to address this question. In recent years a number of large initiatives have been gathering such data: the Dominantly Inherited Alzheimer Network (DIAN) study, a multicentre project established with clinical sites across the USA, Australia and in the UK; and the Alzheimer’s Prevention Initiative (API), which studies a large Colombian kindred affected by the PSEN1 E280A mutation. So far, the results reported from these studies have largely been cross-sectional but as symptoms tend to arise at a similar age within a family, the ages at which an individual’s relatives became clinically affected have been used to predict how far from symptom onset a presymptomatic participant may be. On this basis, amyloid imaging with Pittsburgh compound B (PIB)-PET in DIAN and Florbetapir-PET in API have demonstrated accumulation of Aβ in mutation carriers who are as far as 15 years below their expected age at symptom onset.5-7 This confirms findings from earlier smaller studies, that Aβ deposition on PET is an early event in presymptomatic FAD. A striking observation from the initial FAD PIB-PET studies was that Aβ deposition was most intense in the striatum (Figure 1); a pattern reported for a variety of PSEN1 mutations and for APP mutation and duplication cases.8-10 This striatal predominance does not appear to be seen with the Colombian kindred PSEN1 mutation, and other PSEN1 mutations have subsequently been found to be associated with more prominent thalamic PIB uptake.11 The topography of early amyloid deposition in FAD caused by different mutations therefore appears to be more heterogeneous than was once thought.
Whilst amyloid-PET imaging measures the accumulation of abnormal protein, the other imaging modalities that have been applied to presymptomatic FAD are thought to capture aspects of neurodegeneration further downstream. The most widely used of these have been FDG-PET to measure glucose metabolism and structural MRI to investigate atrophy. MR spectroscopy has also been used and found to demonstrate posterior cingulate metabolic changes in presymptomatic mutation carriers who were on average 10 years younger than the mean age at onset for their family.12 Using FDG-PET, widespread hypometabolism has been observed in presymptomatic FAD in a pattern similar to that seen in sporadic AD.13 In the DIAN study, parietal hypometabolism was evident from around 10 years prior to the parental age at symptom onset. Most regions appeared to show the sequence of events predicted by theoretical biomarker models of AD, with amyloid accumulation occurring first, followed by hypometabolism followed by atrophy14 (Figure 2).
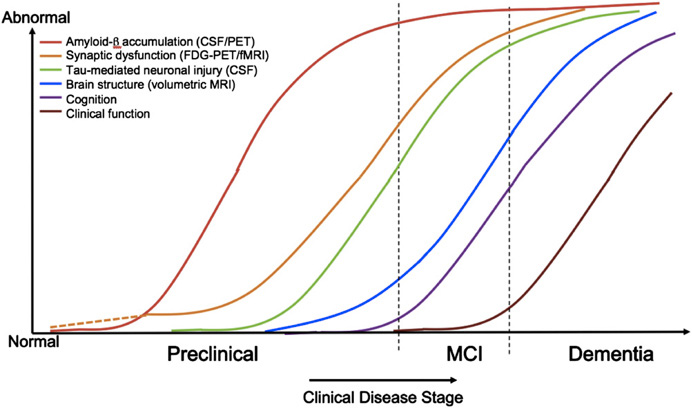
However, the DIAN data also indicated that there may be more complexity to the trajectory of presymptomatic biomarker changes than such models at first suggest. Firstly, different brain regions appear to vary in their vulnerability to the presence of amyloid pathology. Whilst all subcortical regions showed elevated PIB uptake, hypometabolism was only evident in the hippocampus.6 Secondly, the direction of biomarker change was not always as predicted. In very young mutation carriers who were ~25 years from expected age at onset, there was some suggestion that there might be a hypermetabolic phase in some regions including precuneus and posterior cingulate. The numbers in this subgroup were small, however, so replication of the results in a larger cohort will be required. Structural MRI is the imaging modality that has been most thoroughly investigated in FAD and, importantly, it has been used to study mutation carriers longitudinally from a presymptomatic stage up to the actual age at onset of clinical symptoms. Accelerating hippocampal and whole brain atrophy rates have been observed at 5.5 and 3.5 years before symptom onset in mutation carriers.15 Cortical thinning of precuneus and posterior cingulate cortex has been reported approximately four and three years before onset respectively.16 In these small cohort studies, longitudinal measures were able to detect presymptomatic change earlier than when a cross-sectional approach was taken. Visual rating of hippocampal atrophy in presymptomatic FAD appears to be relatively insensitive.17 In view of the early striatal and thalamic amyloid deposition witnessed in FAD, more recent studies have also examined whether these subcortical structures undergo presymptomatic atrophy. We reported decreased volumes of thalamus and caudate in mutation carriers who were on average 5.6 years from parental age at onset, at a stage where hippocampal atrophy was not yet evident.18 Presymptomatic atrophy of thalamus, caudate and putamen has also been reported in a separate cohort of mutation carriers who were ~15 years younger than the median age of dementia diagnosis in their family.19 A voxel-based morphometry analysis of DIAN data demonstrated decreasing thalamic grey matter in presymptomatic DIAN mutation carriers closer to age at onset.20 Subsequent analysis of a larger cohort from DIAN using an automated segmentation technique to assess cortical thickness and subcortical regional volumes demonstrated atrophy of the hippocampus, putamen, thalamus, amygdala and accumbens ~10 years and cortical thinning, particularly of precuneus, ~5 years before the parental age at symptom onset.6 The lack of caudate atrophy in the DIAN data is somewhat surprising, particularly given the high amyloid burden observed in this structure. However, the caudate has been associated with a number of unexpected results in studies of presymptomatic FAD. Lee et al. found that, although caudate volumes were reduced in young presymptomatic mutation carriers, there was a trend towards increasing caudate size in symptomatic mutation carriers during the pre-dementia phase.19 Another group reported increased caudate volumes and cortical thickness in precuneus and parietotemporal areas in a small number of mutation carriers who were approximately a decade younger than their family’s median age at symptom onset – with the intriguing possibility that early amyloid deposition may cause volume increases perhaps due to plaque-associated inflammatory responses.21 Like the report of hypermetabolism in very young mutation carriers, these observations of possible increases in regional volumes require replication in larger studies to ensure that they are not artefactual but they do raise the possibility that biomarkers may change in unexpected ways during the presymptomatic stages of the disease.
Diffusion tensor imaging (DTI) is another modality that has been found to show some unexpected abnormalities in presymptomatic FAD. This advanced MRI technique provides insights into tissue microstructure by examining the magnitude and directionality of water diffusion within white matter tracts and fibre-rich grey matter regions. Diffusion in white matter is described as anisotropic as it occurs preferentially along the major axis of a fibre. The three-dimensional nature of this diffusion is characterised by a diffusion tensor, from which various metrics can be extracted. Fractional anisotropy (FA) describes the overall shape of the tensor, with axial (AxD) and radial diffusivity (RD) representing diffusion in the principal direction of the tract and in the plane perpendicular to this. Mean diffusivity (MD) describes the overall magnitude of diffusion. Early DTI studies focused on white matter FA alone and decreased FA in the fornix, interpreted as loss of structural integrity, was reported in presymptomatic mutation carriers who were on average 13 years younger than the age at dementia diagnosis in their family.22 We recently made a different observation; symptomatic mutation carriers were found to have the expected widespread reductions in white matter FA with increased MD, RD and AxD, however presymptomatic mutation carriers demonstrated a contrasting decrease in MD and AxD in the right cingulum.18 Reductions in AxD occur in axonal injury, which we hypothesised may be an early event in presymptomatic FAD. We also examined diffusion characteristics in grey matter regions of interest and found a corresponding reduction in MD in the right hippocampus. Reduced caudate MD has been reported in another cohort of presymptomatic PSEN1 mutation carriers and it has been suggested that reductions in MD may reflect early pathological responses to amyloid accumulation such as microglial activation and/or swelling of neurons and glia.21 Finally, we observed increased FA in the thalamus and caudate of presymptomatic mutation carriers, which we proposed may be due to the degeneration of long-coursing white matter tracts with preservation of short interneurons within these highly connected structures. Refinement of DTI analysis methods and their application to larger cohorts, particularly those with longitudinal data, will hopefully allow these initial findings to be explored in greater depth.
Another advanced MRI technique that is starting to be applied to the study of presymptomatic FAD is functional MRI (fMRI), which appears to be capable of detecting very early differences between mutation carriers and their mutation-negative siblings. In the Colombian kindred, altered hippocampal activation during memory encoding on fMRI and decreased right parietal volume has been reported approximately two decades before the median age at onset of mild cognitive impairment for the family.23 Another study has found decreased left hippocampal fMRI activity during memory retrieval in mutation carriers.24 In the DIAN study, resting state fMRI data has been analysed to look at functional connectivity in the default mode network; the set of brain regions whose coordinated activity during wakeful rest appears to be critical for successful memory function. Reduced connectivity in precuneus/ posterior cingulate and right parietal cortex was observed in mutation carriers on average 12 years from parental age at onset, with decreasing functional connectivity observed in those closer to their expected age at onset.25
One of the challenges in comparing different imaging studies of presymptomatic AD is that different groups have used different measures to estimate how far from clinical onset the mutation carriers may be. These include years from the parental age at symptom onset, the mean or median age at symptom onset for the family or the mean/median age at diagnosis of mild cognitive impairment or dementia. The latter two of these will of course occur later than the age at symptom onset and may vary according to a range of sociocultural factors surrounding presentation to health professionals. Recollections of when a relative’s symptoms began are themselves subjective and can change over time. Furthermore, although symptoms appear to manifest at broadly similar ages within families, there is variability. Our understanding is currently quite limited regarding both the degree of such variability and the potential genetic or environmental modifiers that may underpin it. All of these caveats should be considered when cross-sectional studies are used to predict the temporal evolution of imaging changes in presymptomatic FAD. Comparability between studies may also be limited by differences in image analysis techniques as variability in methods, for example the approach used to segment a structure of interest like the caudate, may have an influence on the results generated. Finally, it remains uncertain how heterogeneously different FAD mutations affect imaging biomarkers in the presymptomatic phase.
Overall, the literature to date indicates that a variety of imaging biomarker abnormalities is evident in presymptomatic FAD. The use of a range of modalities has started to provide insights into the selective vulnerability of different regions to Aβ pathology, which longitudinal data should help to clarify. Longitudinal imaging will also be important for exploring the suggestion that the direction of change in some biomarkers may be dynamic at different points during the presymptomatic stage of disease. Appreciating the possibility that imaging biomarkers may change in unpredictable ways during preclinical disease is important, particularly in light of upcoming treatment trials. Previous clinical trials have shown that treatments can also have unexpected effects on biomarkers, with the greatest volume losses in the AN1792 Aβ active immunisation trial actually seen in antibody-responders.26 As the API and DIAN study have now both launched presymptomatic prevention trials of amyloid-modifying therapies, information about the sequence of biomarker changes during the natural history of the disease and in the context of treatment will be acquired in tandem. Study of the asymptomatic at-risk state of sporadic AD may progress in a similar fashion, with plans underway for an anti-amyloid treatment trial in healthy older people with evidence of Aβ deposition on amyloid PET scans (the A4 trial). In this context, where imaging will be used to both define and study an at-risk state, a multi-modal approach will be important and insights from the study of presymptomatic FAD may be particularly valuable.
References
- Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. AlzheimersDement 2011;7:280-92.
- Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. The New England journal of medicine 2014;370:311-21.
- Karran E, Hardy J. Antiamyloid therapy for Alzheimer’s disease–are we on the right road? The New England journal of medicine 2014;370:377-8.
- Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 2010;9:1118-27.
- Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. The New England journal of medicine 2012;367:795-804.
- Benzinger TL, Blazey T, Jack CR, Jr., et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America 2013;110:E4502-9.
- Fleisher AS, Chen K, Quiroz YT, et al. Florbetapir PET analysis of amyloid-beta deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: a cross-sectional study. Lancet Neurol 2012;11:1057-65.
- Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci 2007;27:6174-84.
- Remes AM, Laru L, Tuominen H, et al. Carbon 11-labeled pittsburgh compound B positron emission tomographic amyloid imaging in patients with APP locus duplication. Arch Neurol 2008;65:540-4.
- Villemagne VL, Ataka S, Mizuno T, et al. High striatal amyloid beta-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol 2009;66:1537-44.
- Knight WD, Okello AA, Ryan NS, et al. Carbon-11-Pittsburgh compound B positron emission tomography imaging of amyloid deposition in presenilin 1 mutation carriers. Brain 2011;134:293-300.
- Godbolt AK, Waldman AD, MacManus DG, et al. MRS shows abnormalities before symptoms in familial Alzheimer disease. Neurology 2006;66:718-22.
- Mosconi L, Sorbi S, de Leon MJ, et al. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J NuclMed 2006;47:1778-86.
- Jack CR, Jr., Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 2010;9:119-28.
- Ridha BH, Barnes J, Bartlett JW, et al. Tracking atrophy progression in familial Alzheimer’s disease: a serial MRI study. Lancet Neurol 2006;5:828-34.
- Knight WD, Kim LG, Douiri A, Frost C, Rossor MN, Fox NC. Acceleration of cortical thinning in familial Alzheimer’s disease. Neurobiol Aging 2011;32:1765-73.
- Ringman JM, Pope W, Salamon N. Insensitivity of visual assessment of hippocampal atrophy in familial Alzheimer’s disease. J Neurol 2010;257:839-42.
- Ryan NS, Keihaninejad S, Shakespeare TJ, et al. Magnetic resonance imaging evidence for presymptomatic change in thalamus and caudate in familial Alzheimer’s disease. Brain 2013;136:1399-414.
- Lee GJ, Lu PH, Medina LD, et al. Regional brain volume differences in symptomatic and presymptomatic carriers of familial Alzheimer’s disease mutations. J Neurol Neurosurg Psychiatry 2013;84:154-62.
- Cash DM, Ridgway GR, Liang Y, et al. The pattern of atrophy in familial Alzheimer disease: volumetric MRI results from the DIAN study. Neurology 2013;81:1425-33.
- Fortea J, Sala-Llonch R, Bartres-Faz D, et al. Increased cortical thickness and caudate volume precede atrophy in PSEN1 mutation carriers. JAlzheimersDis 2010;22:909-22.
- Ringman JM, O’Neill J, Geschwind D, et al. Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer’s disease mutations. Brain 2007;130:1767-76.
- Reiman EM, Quiroz YT, Fleisher AS, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol 2012;11:1048-56.
- Braskie MN, Medina LD, Rodriguez-Agudelo Y, et al. Memory performance and fMRI signal in presymptomatic familial Alzheimer’s disease. Human brain mapping 2013;34:3308-19.
- Chhatwal JP, Schultz AP, Johnson K, et al. Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology 2013;81:736-44.
- Fox NC, Black RS, Gilman S, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 2005;64:1563-72.