





Summary
We present a case of cerebral venous sinus thrombosis (CVT) caused by paroxysmal nocturnal haemoglobinuria (PNH). Although a rare cause of a rare condition, PNH can be diagnosed with a single blood test and has a poor prognosis if untreated. However therapy with the anti-complement drug, eculizumab, is very effective treatment. Eculizumab usually prevents further thrombotic problems, meaning that early diagnosis is critical. Our approach to investigating the cause of CVT has changed as a result of this case and we believe that PNH is a condition that neurologists dealing with CVT should be aware of.
An elderly patient experienced a sudden onset of visual problems characterised by an inability to send a text message on their mobile phone, set the burglar alarm at their house or read the newspaper. The episode was associated with a mild, throbbing, bi-temporal headache that reached maximum intensity over a couple of minutes. The headache lasted for 30 minutes before resolving spontaneously but vision did not improve. The patient arranged to visit the opticians a few days later. Bilateral papilloedema was identified and she was urgently referred into hospital.
Past medical history included a portal vein thrombosis diagnosed in 2006 which lead to the development of non-cirrhotic portal hypertension. In 2008 the patient became anaemic without an underlying cause identified. At the same time they had an episode of haematemesis secondary to oesophageal varices caused by portal hypertension. To help reduce portal venous pressures the patient underwent a transjugular intrahepatic portal-systemic shunt (TIPSS) insertion and remained under hepatology follow up. The patient developed further thromboses within the TIPSS in 2010 and underwent a TIPSS thrombectomy and further stent insertion. In 2011 recanalisation was attempted via clot retrieval and thrombolytic therapy. This thrombolysis treatment was successful but the TIPSS clotted again in March of 2012. Other medical history included poliomyelitis as a child.
Examination in hospital revealed bilateral reduced visual acuity to 6/12 with normal visual fields to confrontation and normal pupillary reactions. Inspection of the fundi confirmed bilateral grade 4 papilloedema. The remainder of the neurological examination did not demonstrate any new focal neurology and general physical examination was normal.
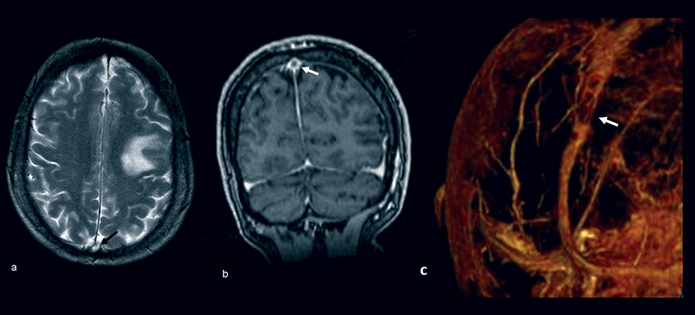
An MRI brain scan and MR venogram showed an acute posterolateral frontal lobe infarction and demonstrated filling defects in the mid and posterior aspect of the superior sagittal sinus (Figure 1 below). A diagnosis of venous cortical infarct and venous sinus thrombosis was made. Thrombophilia screen testing for antithrombin III, protein C, activated protein C resistance, protein S, lupus anticoagulant, prothrombin gene mutation, factor V Leiden mutation and anticardiolipin antibodies were normal.
Treatment was commenced with low molecular weight heparin and then converted to oral anticoagulation with warfarin. The patient’s visual acuity did not improve and remained at 6/12 bilaterally. A further admission to hospital occurred one month after discharge, because the headache returned, but no new abnormality was identified on repeat imaging. During that admission, results of a test for PNH that had been performed by the hepatologists at an out-patient clinic appointment the previous week demonstrated 93.33% of the patient’s cells were PNH. The patient was urgently reviewed by the haematologists and commenced treatment with eculizumab, a monoclonal antibody that inhibits the formation of terminal complement.
Discussion
Cerebral venous sinus thrombosis is an uncommon condition thought to affect approximately five people per million annually.1 Although there are over 100 different aetiologies for CVT, in 12.5-20% of cases an underlying explanation cannot be found.2,3 CVT causes can be divided into acquired risks (such as cancer, pregnancy, oral contraceptive use) and genetic risks (such as the inherited thrombophilias).4 The International Study on Cerebral Vein and Dural Sinus Thrombosis (ISCVT), the largest prospective observational study, included 624 adult cases of CVT and found that thrombophilia was the most common risk factor, seen in 34% of patients.5
PNH is an extremely rare condition with an incidence of around 1.3 per million of the population.6 It is caused by somatic mutations of the PIG-A gene, one of a number of genes needed for the synthesis of the glycophosphatidylinositol (GPI) anchor. The GPI anchor is made in the endoplasmic reticulum and then transported to the surface of the blood cell where it is needed by a variety of cell surface proteins for them to be expressed.7 Lack of function of the GPI protein leads to the absence or reduced expression of key cell surface molecules on blood cells. This renders the cells susceptible to complement mediated attack which causes intravascular haemolysis and platelet activation. This intravascular haemolysis is the hallmark of the disease and as well as being anaemic, patients can experience symptoms including haemoglobinuria, dysphagia, recurrent abdominal pain, dyspnoea, severe lethargy and renal impairment. Classically haemoglobinuria is worse in the morning. Urine colour can change dramatically throughout the day (Figure 2). However, not all patients with PNH describe this symptom so the disease should still be suspected.

PNH causes an increase in mortality as well as morbidity. A study on a British population revealed that patients with PNH, with an average age at presentation of 42 years, treated with supportive measures only had a median survival of only 10 years.8 The main cause of death in PNH is thrombosis with up to 39% of patients developing thromboemboli at some point.8 Once an individual develops a thrombosis their risk of dying is increased by between 5 to 15 times.8,9 Anticoagulation once a thrombosis has occurred is often ineffective. Thromboses in people with PNH, which may be arterial as well as venous, often develop in unusual sites, such as cerebral, mesenteric, hepatic and portal veins.10
Testing for PNH involves sending a blood sample for flow cytometry.7,11 Flow cytometry for the GPI-linked proteins is available through most laboratories around the UK. Once a diagnosis is made patients should be referred to one of the two nationally commissioned PNH centres.These are based in St James’s University Hospital in Leeds or Kings College Hospital in London.
Treatment of PNH was predominantly supportive until the monoclonal antibody, eculizumab, was licensed in 2007. Eculizumab binds to the complement protein C5, arresting the complement cascade and therefore preventing terminal complement activation and haemolysis of red cells.7 Clinical trials have shown that eculizumab dramatically decreases the risk of venous-thromboembolism.10 Although not a cure, patients with PNH who are treated with eculizumab have survival rates comparable with age and sex-matched normal controls.12 Blocking the cleavage of the C5 protein increases the risk of developing infections with Neisseria Meningitidis and so vaccination is required prior to starting treatment.7
International experts in PNH suggest this disease should be tested for in all cases of CVT.11 We now test for PNH in cases of unexplained CVT, in cases where there is a past history of venous thrombosis and where there is evidence of anaemia, other cytopenia or intravascular haemolysis.
PNH is a rare but important condition that neurologists should consider when managing patients with CVT. Thrombosis in the setting of PNH is an urgent indication to commence eculizumab through the National PNH Service. Before this case PNH did not enter our differential diagnosis. The poor outcomes seen in untreated PNH, and the significant improvements once treated with eculizumab, makes it a diagnosis not to be missed.
References
- Bousser MG, Ferro JM. Cerebral venous thrombosis: an update. Lancet Neurol 2007; 6:162-70
- Masuhr F, Mehraein S, Einhäupl K. Cerebral venous and sinus thrombosis. J Neurol 2004; 251: 11-23
- Kimber J. Cerebral venous sinus thrombosis. Q J Med 2002; 95: 137-142
- Saposnik G, Barinagarrementeria F, Brown RD Jr et al. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011; 42: 1158-92
- Ferro JM, Canhão P, Stam J et al. Prognosis of cerebral vein and dural sinus thrombosis: results of the International Study on Cerebral Vein and Dural Sinus Thrombosis (ISCVT). Stroke 2004; 35: 664-70
- Hill A, Platts PJ, Smith A et al. The incidence and prevalence of paroxysmal nocturnal haemoglobinuria (PNH) and survival of patients in Yorkshire (Abstract). Blood 2006; 108: 985.
- Kelly R, Richards S, Hillmen P, Hill A. The pathophysiology of paroxysmal nocturnal hemoglobinuria and treatment with eculizumab. Ther Clin Risk Manag 2009; 5: 911-21.
- Hillmen P, Lewis SM, Bessler M et al. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med 1995 ; 333: 1253-8.
- de Latour RP, Mary JY, Salanoubat C, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood 2008: 112:3099-106.
- Hillmen P, Muus P, Dührsen U et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood 2007; 110: 4123-8
- Parker C, Omine M, Richards S et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005: 106: 3699-709
- Kelly RJ, Hill A, Arnold LM et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood 2011; 117: 6786-92.