

Summary
- Prion diseases are characterised by the accumulation of misfolded prion protein (PrP) and widespread neuronal loss throughout the brain. Recent work has elucidated a major mechanism by which misfolded PrP induces neurodegeneration in prion disease.
- Rising levels of misfolded PrP lead to sustained dysregulation of an endogenous cellular pathway, the unfolded protein response (UPR), which regulates protein synthesis at the level of translation initiation.
This results in the sustained reduction in global protein synthesis rates in neurons, leading to loss of critical proteins, resulting in synaptic failure and neuronal death. - Genetic and pharmacological manipulation of this pathway to restore protein synthesis prevents neuronal loss, reverses cognitive deficits and abrogates clinical disease.
- The same branch of the UPR is over-activated in other protein misfolding disorders, including Alzheimer’s and Parkinson’s diseases, ALS and PSP. Further, it is a key pathway in learning and memory. Therefore, UPR modulation to restore protein synthesis levels in neurons is potentially an important new therapeutic strategy for neurodegenerative disease.
Background
Prion diseases are rare neurodegenerative disorders that belong to the emerging group of protein misfolding diseases, which includes Alzheimer’s and Parkinson’s diseases. In each case, the accumulation of a disease specific protein is associated with a relatively stereotyped clinicopathological syndrome. How neuronal loss occurs in these disorders is not clear, but recent work has revealed the mechanism by which protein misfolding leads to neurodegeneration in prion disease.
The central pathogenic process in prion disorders is the formation and accumulation of an aberrantly folded conformer (PrPSc) of the host-encoded cellular prion protein (PrPC).1 PrPSc is generated from PrPC through an autocatalytic post-translational change in secondary structure (Figure 1).2 The misfolded protein aggregates and accumulates throughout the brain, is accompanied by astrocytosis, spongiform change and extensive neuronal cell loss. Whilst PrPSc is associated with infectivity (prion diseases are transmissible), there is extensive evidence that it is not in itself neurotoxic. Sub-clinical states of prion disease have been identified in which extensive accumulation of PrPSc is dissociated from neurotoxicity.3 PrPSc is harmless to cells devoid of PrPC, and therapeutic agents targeting PrPSc have very limited efficacy and do not prevent neuronal loss. PrPC is absolutely required for susceptibility to prion neurotoxicity: PrP-null mice are resistant to prion disease4 and depleting PrPC in neurons of prion infected mice cures disease, as conversion can no longer occur.5 Thus, the process of prion protein misfolding is central to neurotoxicity. Recent work has shown that neuronal death results from dysregulation of the cellular response to unfolded proteins triggered by the process of prion protein misfolding.6
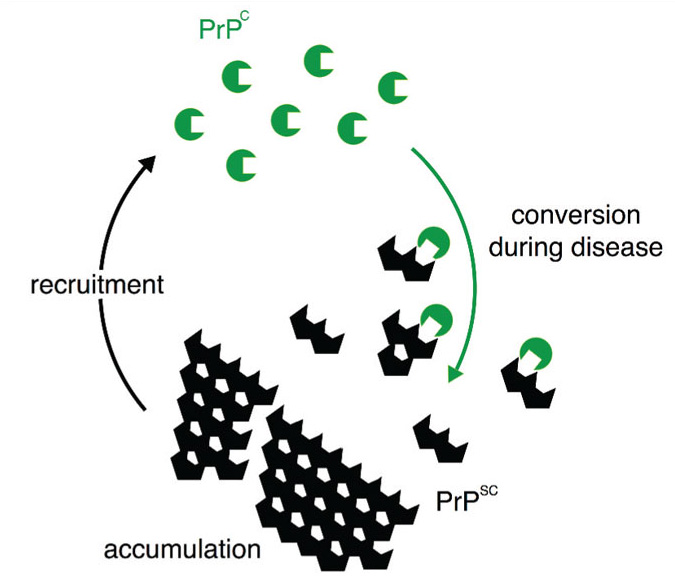
Native prion protein (PrPC; green partial circles) is converted into PrPSc (black partial hexagons) in an autocatalytic process during prion replication. It is likely that PrPSc seeds (multimers) interact with individual PrPC molecules. The two proteins have identical primary structure but differ in secondary structure: PrPC is predominantly alpha helical whereas PrPSc is rich in beta-sheet, protease resistant and accumulates, recruiting more PrPC for further cycles of conversion.
The unfolded protein response
All cells need correctly folded proteins for normal functioning. The build up of unfolded proteins within the endoplasmic reticulum (ER) constitutes a form of cellular stress that elicits a protective signalling cascade, the Unfolded Protein Response (UPR), which maintains protein-folding homeostasis, “proteostasis”.7 Rising levels of misfolded proteins in the ER are detected by Binding immunoglobulin protein (BiP), which results in activation of the three branches of the UPR to increase protein folding through chaperone expression (via ATF6 and IRE1 branches) and to transiently reduce protein levels by inhibiting protein synthesis (PERK branch). This occurs via the phosphorylation first of PERK and then of the alpha subunit of eIF2 (eIF2α), which is needed for formation of ternary complex and initiation of translation. Activation of the UPR is usually a transient event that terminates when eIF2α-P is dephosphorylated by GADD34, rapidly restoring protein translation.8
The UPR in prion disease
Recent work in prion diseased mice revealed that rising levels of misfolded prion protein caused and sustained increase in the phosphorylation of PERK and eIF2α in neurons.6 The effect of this is the sustained reduction in global protein synthesis rates in neurons, causing catastrophic decline in levels of key proteins including synaptic proteins, vital for healthy functioning and neuronal survival. The result is neurodegeneration. Genetic manipulation of the pathway to reduce levels of eIF2α-P restored vital protein synthesis rates and was profoundly neuroprotective (Figure 2). This was true for upstream (knockdown of PrP levels) or downstream (by lentivirally mediated overexpression of the eIF2α-P phosphatase GADD34) interventions that reduced eIF2α-P levels. The resultant rescue of protein synthesis rates prevented decreases in synaptic protein levels, maintained synapse number and synaptic function, preventing behavioural and cognitive deficits, and resulted in extensive neuroprotection. Most importantly, there was a significant increase in survival. Notably, inhibiting the dephosphorylation of eIF2α-P with the small molecule salubrinal, had the opposite effect, exacerbating the decrease in protein synthesis and accelerating disease. Critically, this rescue occurs downstream of prion replication and independently of it. PrPSc levels are unaffected.
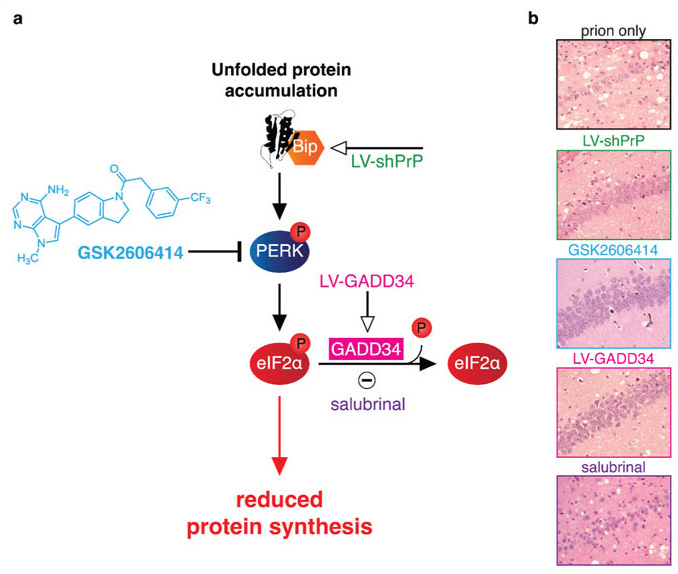
Pharmacological modulation of the UPR in prion disease
These data led to the prediction that pharmacological inhibition of PERK- eIF2α-P signalling would be similarly neuroprotective. GSK2606414, a highly selective PERK inhibitor,9 was orally administered to prion-infected mice daily from points both before and after the onset of behavioural deficits.10 PERK inhibition by GSK2606414 similarly prevented elevated levels of eIF2α-P and decline in protein synthesis rates and resulted in extensive neuroprotection throughout the brain. Encouragingly, treatment that was started even after the emergence of cognitive deficits had the same beneficial effects as treatment from earlier time-points. This work was the first description of a small molecule able to prevent neuronal loss and clinical disease in vivo.
Wider relevance
There is increasing evidence that UPR dysregulation is a central process in protein misfolding neurodegenerative diseases, and that maintaining translation levels is essential for neuronal health. Increased PERK-P and eIF2α-P have been reported in post-mortem analyses of brains of patients with AD, PD, ALS, and the tauopathy progressive supranuclear palsy (PSP), as well as prion disease.11 Genetic polymorphisms in PERK pre-dispose to PSP.12 The pathway is also implicated in learning and memory; manipulation of eIF2α-P levels boost cognition in wild type mice and restore memory deficits in AD mouse models. Thus, it would appear that targeting UPR dysregulation that occurs downstream of misfolded protein replication, be this prion, amyloid beta, tau, α-synuclein or TDP-43 may hold promise for new treatments for neurodegenerative disorders more broadly.
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982;216(4542):136-44.
- Prusiner SB. Creutzfeldt-Jakob disease and scrapie prions. Alzheimer Dis Assoc Disord 1989;3(1-2):52-78.
- Hill AF, Collinge J. Subclinical prion infection in humans and animals. Br Med Bull 2003;66:161-70.
- Bueler H et al. Mice devoid of PrP are resistant to scrapie. Cell 1993;73(7):1339-47.
- Mallucci GR et al. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 2007;53(3):325-35.
- Moreno JA et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 2012;485(7399):507-11.
- Ron D,. Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007;8(7): 519-29.
- Novoa I et al. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 2001;153(5):1011-22.
- Axten JM et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 2012;55(16):7193-207.
- Moreno JA et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 2013;5(206):206ra138.
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 2014;15(4):233-49.
- Stutzbach LD et al. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun 2013;1(1):31.