


Inherited disorders of the peripheral nerve may be classified into those in which neuropathy is the main feature, and those in which it is part of a more generalised disorder. The advent of novel sequencing technologies has started to revolutionise the screening of known genes and allowed the identification of numerous genes to be newly associated with neuropathies, including Charcot.Marie.Tooth disease (CMT), distal hereditary motor neuropathy (dHMN), hereditary sensory and autonomic neuropathy (HSAN) and hereditary neuralgic amyotrophy (HNA). Indeed, the past two years have seen a significant shift from traditional Sanger sequencing to next generation sequencing (NGS) methods including whole genome sequencing, exome sequencing and customised gene panels. As a result, the number of peripheral neuropathy-related genes has nearly doubled in the past few years and continues to rise. This review will provide an update on the genetics of the inherited neuropathies and the implications of NGS to both research and diagnostic services.
Charcot-Marie-Tooth Disease, hereditary sensory and autonomic neuropathy, distal hereditary motor neuropathy and hereditary neuralgic amyotrophy CMT, otherwise known as hereditary motor and sensory neuropathy (HMSN) is a heterogeneous group of diseases, both clinically and genetically. Patients typically exhibit wasting and weakness of distal muscles, reduced reflexes, foot deformities (pes cavus), sensory loss and walking difficulties. CMT is the most common inherited neuromuscular disease, with a prevalence of approximately 1 in 2500, with some variation between populations.1 In the era prior to the identification of the chromosome 17 duplication including the peripheral myelin protein 22 (PMP22) gene in CMT patients in the 1990s,2,3,4,5 CMT was categorised primarily according to neurophysiological studies and peripheral nerve pathology.6 Patients with upper limb motor nerve conduction velocities (NCVs) under 38m/s are classified as CMT1, or demyelinating CMT, and patients with NCVs greater than 38m/s as CMT2, or axonal CMT. An evermore recognised intermediate form describes patients with both axonal and demyelinating features and NCVs between 25 and 45 m/s.7 The advances made in the genetics of neuropathies in the past two decades have further subdivided CMT according to the pattern of inheritance and over 40 causative genes or loci.6 Autosomal recessive (AR) forms of CMT1 are referred to as CMT4. CMT3 (also called congenital hypomyelinating neuropathy (CHN) or Dejerine-Sottas disease (DSD)) is sometimes used to classify children with a severe demyelinating or hypomyelinating neuropathy usually associated with de novo dominant mutations in PMP22, myelin protein zero (MPZ) or early growth response 2 (EGR2).8
Genes involved in CMT encode proteins with increasingly diverse roles, including myelin structural components, transcription factors needed for gene regulation, protein synthesis, axonal transport, endocytosis and protein sorting, mitochondrial fusion and fission, and ion channels. Metabolic enzymes have also been implicated in CMT: mutations in PRPS1 have been associated with hereditary peripheral neuropathy with hearing loss and optic neuropathy (CMTX5), opening up the possibility of treating this CMT subtype with antimetabolite therapy.9
HSAN is caused by the degeneration of sensory and autonomic neurons. Patients often present with sensory loss and ulcerative mutilations. Autonomic involvement is more likely in AR forms, while motor problems tend to be associated with AD forms of HSAN. AR forms usually have earlier onset. HSAN is categorised into five subtypes according to age of onset, inheritance and phenotype. The 12 genes implicated in HSAN thus far encode proteins with roles in sphingolipid metabolism, DNA methylation, endoplasmic reticulum tubulation, membrane excitability, cytoskeletal organisation, axonal guidance during development and vesicular transport.10 In contrast, dHMN, also known as distal spinal muscular atrophy (dSMA) predominantly involves motor nerves although some degree of sensory problems may be observed. dHMN is classified into seven subtypes; both AD and AR inheritance have been described. Functions of proteins implicated in dHMN include chaperones, protein synthesis, RNA and DNA unwinding, axonal transport, ion channels and metallation of copper enzymes.11
Novel findings in the genetics of peripheral neuropathies
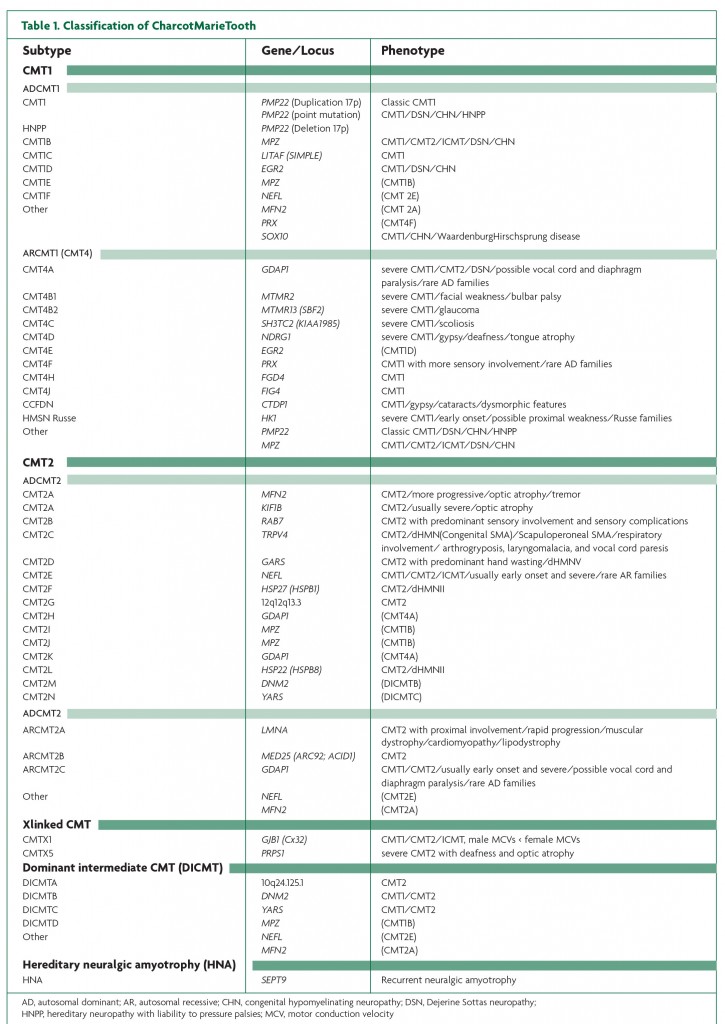
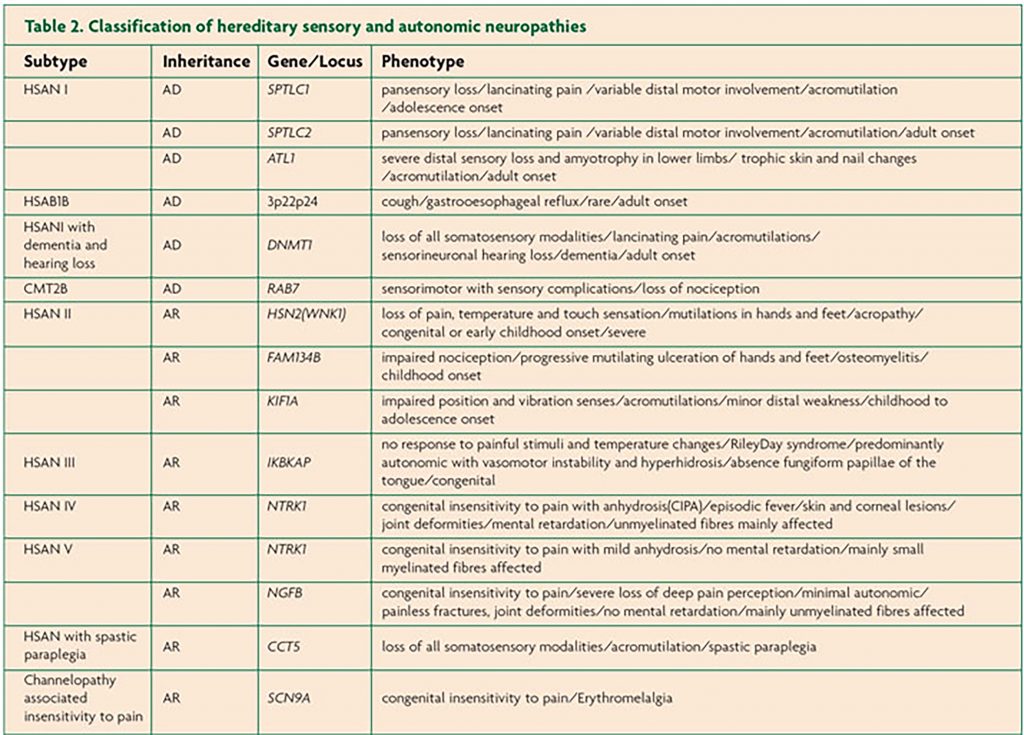
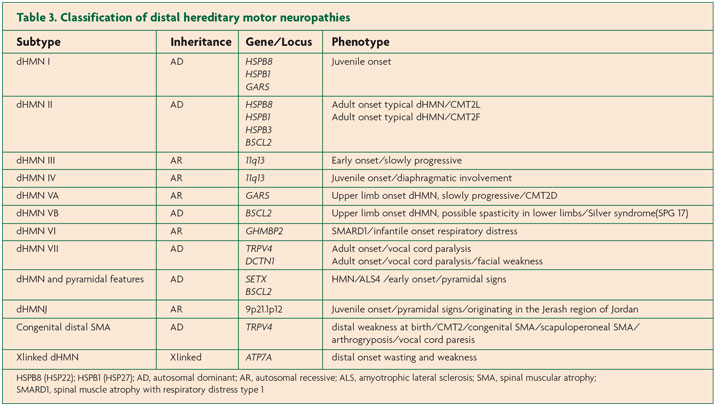
The list of genes implicated in inherited peripheral neuropathies is constantly growing. Tables 1, 2 and 3 summarise the genetic subtypes and associated phenotypes in CMT, HSAN and dHMN.
In addition to the identification of new genes, unexpected mutations in previously known peripheral neuropathy genes have been discovered. An alternatively sized duplication upstream of PMP22 was detected in a CMT patient; the duplication did not encompass PMP22 and therefore would have been missed by traditional diagnostic methods. The duplicated segment likely contains a regulatory region affecting PMP22 expression.12 In fact, distal enhancers upstream of PMP22 have recently been identified.13 A shorter, 800kb duplication encompassing PMP22 was found in a patient with demyelinating CMT.14 Copy number variants (CNVs) in the PMP22 region arising from new mechanisms involving non-recurrent rearrangements have also been associated with HNPP and CMT1A.15
Besides the chromosome 17 duplication16,17,18 CNVs and whole gene deletions of GJB1, were not thought to be extensively involved in the aetiology of CMT.14 Recently, however, a duplication of the MPZ gene was found to cause CMT. This finding suggests that CNVs may indeed play a role in CMT.19
A synonymous change activating a cryptic splice site in MPZ was found in a patient with DSD.20 Silent nucleotide changes are usually ignored in the analysis of sequencing data. Additional precautions will now need to be taken when categorising such variants as non-pathogenic. Similarly, mutations in the untranslated region upstream of gap junction beta.1 (GJB1) thought to affect splicing have been found in X-linked CMT.21,22
These unexpected findings raise the possibility of novel mechanisms of disease and suggest that new considerations need to be taken into account when interpreting results both in diagnostic and research settings.
Expanding genotype-phenotype correlations
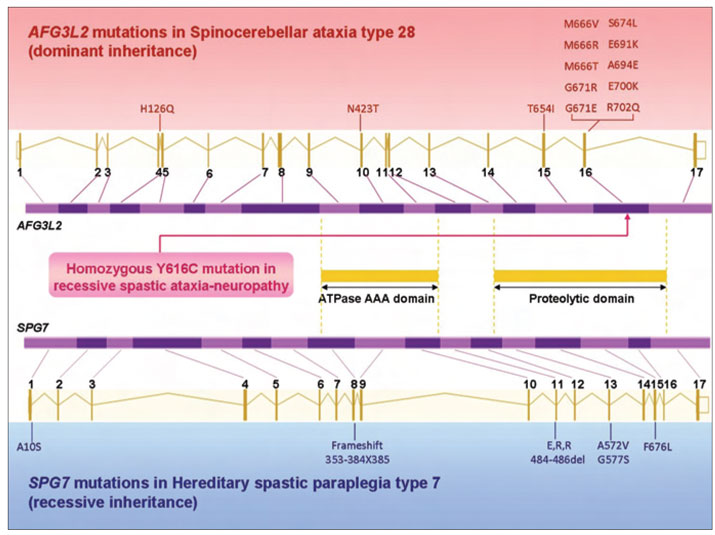
The identification of new peripheral neuropathy genes and new mutations in known peripheral neuropathy genes is leading to increasingly complex genotype-phenotype correlations. Mitochondrial DNA polymerase. (POLG1) mutations were found in a patient with AR axonal CMT associated with tremor and ataxia; however, this patient showed no features commonly associated with mitochondrial disease.23 Similarly, ATP7A mutations, which typically cause severe Menkes disease or occipital horn syndrome,24 were found in a patient with X-linked dHMN and no copper deficiency.25 Mutations in Atlastin-1 (ATL1) were described in a patient with HSAN Type 126; this gene is also associated with early onset hereditary spastic paraplegia SPG3A.27 The genetic basis of a syndrome including AR early onset spastic ataxia and peripheral neuropathy has recently been attributed to mutations in AFG3L2 using exome sequencing.28 AD-inherited mutations in this gene are usually associated with spinocerebellar ataxia type 28 (SCA28).29 This new clinical syndrome had overlapping features of both AD SCA28 and AR hereditary spastic paraplegia type 7,both of which are frequently associated with peripheral neuropathy. Interestingly, both diseases are due to mutations in genes encoding subunits of a particular class of mitochondrial proteases; although these two proteins interact, they lead to strikingly different phenotypes when dysfunctional (Figure 1). This finding also illustrates how NGS can be effective in determining the cause of a complex neuropathy syndrome.28
The causative genes responsible for complex forms of CMT were also recently described; these may represent new syndromes. Mutations in Fibulin-5 (FBLN5), encoding a constituent of the extracellular matrix needed for elastic fibre assembly were found in a case of AD CMT. Mutations were subsequently identified in other CMT families with hyperelastic skin, and in patients with age-related macular degeneration, most of whom had a mild to severe peripheral neuropathy.30 DNA (cytosine-5-)-methyltransferase 1 (DNMT1) mutations were associated with both central and peripheral neurodegeneration in one form of hereditary sensory and autonomic neuropathy with dementia and hearing loss.31 Sporadic or AD intermediate CMT associated with focal segmental glomerulosclerosis (FSGS) was found to be caused by mutations in the inverted formin, FH2 and WH2 domain containing (INF2) gene, encoding a protein known to interact with other proteins essential for myelination and myelin maintenance.32 The association of these genes with such complex phenotypes will help provide new insights into peripheral nerve function.
Frequency of genetic subtypes and diagnostic guidelines
In the European/UK and US population, the AD forms of CMT1 and CMT2 are more common and represent about 90% of CMT patients; however, approximately 40% of CMT patients exhibit AR inheritance in populations with high rates of consanguineous marriage such as the region of the Mediterranean basin.6 The most frequent subtypes of CMT include CMT1A (55% of CMT and 66.8% of CMT1), CMT1X (15.2% of CMT and 18.4% of CMT1), HNPP (9.1% of CMT), CMT1B (8.5% of CMT and 10.4% of CMT1) and CMT2A (4% of CMT and 21.9% of CMT2), while other subtypes account for less than 1% of all CMT.33-37 SH3 domain and tetratricopeptide repeats 2 (SH3TC2) mutations accounted for most of the AR CMT cases (42.9% of CMT4).35 Similar frequencies were obtained in a cohort of CMT patients in Norway, where the most common subtypes were CMT1A followed by CMT1X, CMT2A and CMT1B.38 Serine palmitoyltransferase, long chain base subunit 1 (SPTLC1) mutations account for the majority of AD HSAN cases.39
Considering the number of genes found to be associated with CMT, a diagnostic algorithm encompassing the patient’s ethnic background, neurophysiology, pattern of inheritance, and any outstanding features should prove to be extremely useful to clinicians and should help focus the genetic tests to be performed. The use of these algorithms has enabled 70% of CMT patients to receive a genetic diagnosis.33 Because over half of CMT2 cases remain genetically unexplained39 fewer patients with CMT2 receive a genetic diagnosis compared to CMT1. Pinpointing the disease-causing gene is essential from the clinician and patient’s perspective for improved diagnosis and counselling, as well as for understanding the pathomechanisms underlying the neuropathy and developing targeted treatments.6,40 A genetic diagnosis also helps avoid unnecessary tests and inappropriate therapy trials.8
If the nerve conduction studies indicate demyelination and the phenotype is that of classic CMT with AD inheritance, the chromosome 17 duplication should be tested first. If there is no male-to-male inheritance, GJB1 should be screened; if there is male-to-male transmission, MPZ should be tested, followed by PMP22, and finally the less common genes including EGR2, lipopolysaccharide-induced TNF factor (LITAF) and neurofilament light (N.FL) subunit.6,41
If the neuropathy is AD and axonal with a classic CMT2 phenotype, GJB1 should be screened if there is no male-to-male inheritance,as well as MFN2,especially if the disease is severe with childhood-onset and/or optic atrophy is present. The next genes to be screened in AD axonal neuropathies with a classical phenotype are MPZ (especially for late-onset CMT2), followed by N-FL, alanyl-tRNA synthetase (AARS), ganglioside-induced differentiation-associated protein 1 (GDAP1) and transient receptor potential cation channel, subfamily V, member 4 (TRPV4).6
If the patient presents with AD CMT2 with strong sensory involvement, SPTLC1 should be screened, followed by RAB7. If on the contrary, the neuropathy is AD CMT2 with motor predominance, Berardinelli-Seip congenital lipodystrophy 2 (BSCL2) and glycyl-tRNA synthetase (GARS) should be tested in patients where the upper limbs are mostly affected, and heat shock protein beta.1 (HSPB1), heat shock protein beta.8 (HPSB8), BSCL2 and TRPV4 should be screened in this order for lower-limb predominance.6,34 The overlap between CMT, HSAN and HMN further complicates diagnosis; for example, mutations in two genes associated with either HSAN or CMT can cause a similar phenotype, and mutations in one gene can be implicated in both dHMN and axonal CMT.39
Selecting which genes to screen in AR CMT will depend on the particular phenotype and ethnic background of the patient.6 Genes to be tested in dHMN cases should be prioritised according to inheritance. In AD cases, phenotypic features such as upper versus lower limb onset, vocal cord palsy and pyramidal signs may be used to focus genetic testing.11
Genetic modifiers of disease
Recent years have seen increased interest in identifying potential genetic modifiers of various diseases including hereditary neuropathies. Indeed, the high intra- and inter-familial variability in disease severity typical of CMT1A and of many other subtypes of CMT, HSAN and dHMN renders it difficult for clinicians to advise patients on the way their disease is likely to progress and how severely their children may be affected. A recent study has found that the mRNA levels of certain lipid metabolism genes in the skin biopsies of CMT1A patients could account for 47% of the variance in disease severity.42 The identification of such genetic modifiers will help provide a more accurate disease prognosis, and will likely help understand the disease pathways.
Next-generation sequencing in peripheral neuropathies
Non-Sanger-based sequencing technologies are revolutionising gene discovery and diagnostics for Mendelian and complex diseases. Although traditional ‘first-generation’ Sanger-sequencing and linkage studies are likely to remain important tools in genetic research, we are progressively moving to the era of next-generation sequencing (NGS), which encompasses whole-genome sequencing, exome sequencing and targeted re-sequencing of regions of interest.
All NGS technologies involve preparation and fragmentation of a DNA library, amplification of the library and massively-parallel sequencing of amplicons. The principal differences between these technologies are the enrichment method used and the length of DNA fragments which can be read depending on the sequencing chemistry used. The Roche 454 pyrosequencingTM , Illumina/SolexaTM Genome Analyzer, Invitrogen Ion TorrentTM and Applied Biosystems SOLIDTM system are popular sequencing platforms used in NGS.43,44
High-throughput and cost-efficient sequencing are distinguishing features of next-generation techniques. Other advances include the ability to sequence many bases using 1.2ug of DNA and the ability to investigate disease genes in small families. Next-generation technologies are essential to utilise in heterogeneous disorders such as the inherited peripheral neuropathies. For example, whole-genome sequencing has been used to identify a mutation in a known CMT gene, SH3TC2, in a patient with AR CMT. The authors argue that whole-genome sequencing may be useful in the diagnostic setting for highly penetrant, heterogeneous diseases such as CMT-45 Exome sequencing, which involves target enrichment and high-throughput sequencing of the coding and intronic boundary regions of the genome, was successfully used to identify the causative mutation in a novel gene, dynein cytoplasmic 1 heavy chain 1 (DYNC1H1) in a case of axonal CMT-46 Exome sequencing was also used as a comprehensive diagnostic screen in a CMT patient, leading to the identification of a mutation in a known CMT gene, GJB1-47 Exome sequencing is not only suitable for detecting rare disease-causing variants, but also potential risk factors in protein-coding regions. Moreover, this technique allows causative genes to be found in small families or isolated individuals in whom positional cloning or linkage is not feasible.48
Targeted re-sequencing using customised gene panels is also becoming increasingly popular as it allows researchers to focus on particular areas of interest. This type of NGS is especially well-suited to the diagnostic setting as it involves less data handling and a quicker turnaround time. Panels can be designed for specific diseases or phenotypes, and may include a few genes to hundreds of genes depending on the platform used. The Illumina MiSeq and the Invitrogen Ion TorrentTM are especially suited to such purposes.
Currently, diagnosis by Sanger-sequencing is slow and expensive, and is often not available for many subtypes of inherited neuropathies. Testing of rarer genes is often restricted to the research setting.6 These NGS techniques will undoubtedly change the face of medical diagnostics, including which genes are prioritised for testing49 (Figure 2). Diagnosis of genetically heterogeneous diseases such as inherited neuropathies will become faster and will allow for simultaneous testing of many genes. Although most NGS are currently too expensive to be used routinely, they are quickly becoming more affordable.47 NGS will not only facilitate the identification of new genes, but may also be a tool for finding modifiers of disease severity. Before these techniques are integrated into the clinical setting, extensive knowledge in both molecular biology techniques and bioinformatics will be needed.44
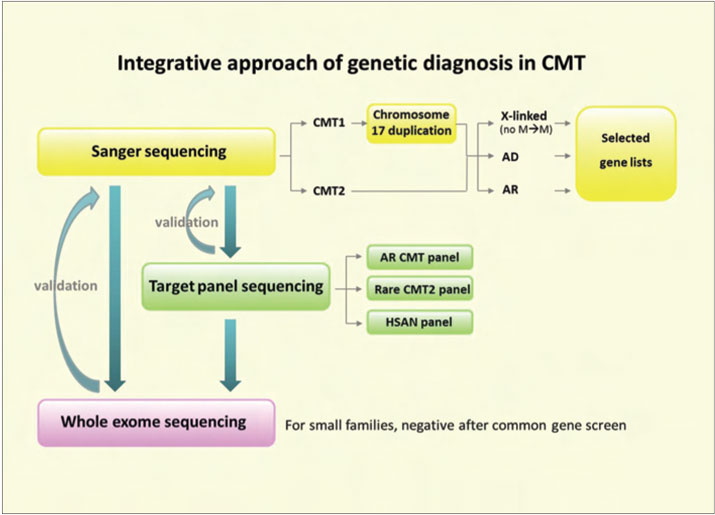
While these NGS methods are more cost-efficient and less time-consuming, it is not advisable to use them routinely without restraint. Unnecessary testing is one of the dangers associated with NGS; genetic testing should be focused and guided by diagnostic algorithms.8,34,50,51 The most common genes should still be tested first, according to inheritance pattern and phenotype47 as mutations in 1 of 4 genes, PMP22, MFN2, MPZ, or GJB1 accounted for over 90% of 527 patients with a known genetic diagnosis of CMT in a recent study by Saporta and colleagues.35
Challenges in Next-Generation-Sequencing and diagnostics
Searching for new genes is an exciting endeavour, but the analysis of the large datasets produced by whole-genome and exome sequencing can be daunting. Although a functional candidate gene approach is advisable, CMT-associated genes are not always nerve-specific and are generally ubiquitously expressed. Recently identified genes have been involved in pathways never previously described to cause inherited neuropathies. It is therefore far from trivial to prepare a list of candidate genes when searching for new causative genes, especially for CMT. Even with these caveats we predict that over the next 18 months, diagnostic exome sequencing will become available in the UK.
Once the list of candidate genes has been reduced to a manageable number, one is still faced with the difficult task of assessing the pathogenicity of the variants. A recent survey of loss-of-function mutations in coding genes has estimated that a single genome from a “healthy” human contains approximately 100 loss-of-function variants.52 This is evident in NGS studies, which produce long lists of potentially pathogenic variants of unknown significance. Whole-genome sequencing studies uncover a high number of variants in coding regions, including in many genes known to cause CMT.53 This issue also applies to traditional Sanger-sequencing methods. Variants previously thought to be pathogenic in CMT patients have later been reclassified as polymorphisms, and vice-versa. Segregation of the mutation in families, frequency in controls, conservation between species and functional studies may help sort through variants.6,54 55 Comprehensive online databases of known variants associated with particular diseases will also be valuable. Good genotype-phenotype studies will also assist clinicians and researchers in interpreting the variants.53
References
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet 1974;6(2):98-118.
- Lupski JR, Oca-luna RMD, Slaugenhaupt S, et al. DNA Duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991;66:219–232. (1)
- Rayemaekers P, Timmerman V, Nelis A, et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT1a). Neuromuscular disord 1991;1(2):93-97. (2)
- Timmerman V, Nelis E, Van Hul W, et al. The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nature 1992;1:171-175. (6)
- Valentijn LJ, Bolhuis PA, Zorn I, et al. The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1A. Nature 1992;1:166-170. (7)
- Reilly MM, Murphy M, Laura M. Charcot-Marie-Tooth disease. J Peripher Nerv Syst 2011;14:1–14. (4)
- Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980;103:259–280. (3)
- Amato AA, Reilly MM. The death panel for Charcot-Marie-Tooth disease panels. Ann Neurol 2011;69(1):1-4.
- Kim H-J, Sohn K-M, Shy ME, et al. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause Hereditary Peripheral Neuropathy with Hearing Loss and Optic Neuropathy (CMTX5). Am J Hum Genet 2007;81:552-558.
- Rotthier A, Baets J, Timmerman V, et al. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat Rev Neurol 2012;8(2):73-85.
- Rossor AM, Kalmar B, Greensmith L, et al. The distal hereditary motor neuropathies. J Neurol Neurosur Ps 2012;83:6-14.
- Weterman MAJ, van Ruissen F, de Wissel M, et al. Copy number variation upstream of PMP22 in Charcot-Marie-Tooth disease. Eur J Hum Genet 2010;18:421-8.
- Jones EA, Brewer MH, Srinivasan R, et al. Distal enhancers upstream of the Charcot-Marie-Tooth type 1A disease gene PMP22. Hum Mol Genet 2012;21(7):1581-91.
- Huang J, Wu X, Montenegro G, et al. Copy number variations are a rare cause of non-CMT1A Charcot-Marie-Tooth disease. J Neurol 2010;257(5):735-741.
- Zhang F, Seeman P, Liu P, et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am J Hum Genet 2010;86:892-903.
- Ainsworth PJ, Bolton CF, Murphy BC, et al. Genotype/phenotype correlation in affected individuals of a family with a deletion of the entire coding sequence of the connexin 32 gene. Hum Genet 1998;103:242–244. 48
- Lin C, Numakura C, Ikegami T, et al. Deletion and nonsense mutations of the connexin 32 gene associated with Charcot–Marie–Tooth disease. Tohoku J Exp Med 1999;188:239–44. 49
- Gonzaga-Jauregui C, Zhang F, Towne CF, et al. GJB1/Connexin 32 whole gene deletions in patients with X-linked Charcot–Marie–Tooth disease. Neurogenetics 2010;11:465-470. 50
- Høyer H, Braathen GJ, Eek AK, et al. Charcot-Marie-Tooth caused by a copy number variation in myelin protein zero. Eur J Med Genet 2011;54(6):e580-e583.
- Taioli F, Cabrini I, Cavallaro T et al. Dejerine-Sottas syndrome with a silent nucleotide change of myelin protein zero gene. J Peripher Nerv Syst 2011;16:59-64.
- Murphy SM, Polke J, Manji H, et al. A novel mutation in the nerve-specific 5’UTR of the GJB1 gene causes X-linked Charcot-Marie-Tooth disease. J Peripher Nerv Syst 2011;16:65-70.
- Kabzinska D, Kotruchow K, Ryniewicz B, et al. Two pathogenic mutations located within the 5’-regulatory sequence of the GJB1 gene affecting initiation of transcription and translation. Acta Biochim Pol 2011;58(3):359-63.
- Harrower T, Stewart JD, Hudson G, et al. POLG1 mutations manifesting as autosomal recessive axonal Charcot-Marie-Tooth disease. Arch Neurol 2008;65(1):133-6.
- Kaler SG, Gallo LK, Proud VK, et al. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat Genet 1994;8:195–202.
- Kennerson ML, Nicholson GA, Kaler SG, et al. Missense mutations in the copper transporter gene ATP7a cause X-linked distal Hereditary Motor Neuropathy. Am J Hum Genet 2010;86:343-52.
- Guelly C, Zhu P-P, Leonardis L, et al. Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of Hereditary Sensory Neuropathy type I. Am J Hum Genet 2011;88:99-105.
- Zhao X, Alvarado D, Rainier S, et al. Mutations in a newly identified GTPase gene cause autosomal dominant Hereditary Spastic Paraplegia. Nat Genet 2001;29:326-31.
- Pierson TM, Adams D, Bonn F, et al. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet 2011;7(10):e1002325.
- Di Bella D, Lazzaro F, Brusco A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet 2010;42:313–321.
- Auer-Grumbach M, Weger M, Fink-Puches R, et al. Fibulin-5 mutations link inherited neuropathies, age-related macular degeneration and hyperelastic skin. Brain 2011;134:1839-52.
- Klein CJ, Botuyan M-V, Wu Y, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 2011;43(6):595-602.
- Boyer A, Nevo F, Plaisier E, et al. INF2 mutations in Charcot–Marie–Tooth disease with glomerulopathy. New Engl J Med 2011;365(25):2377-2388.
- Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot–Marie–Tooth disease. Lancet Neurol 2009;8:654-67.
- Shy ME, Patzkó E. Axonal Charcot-Marie-Tooth disease. Curr Opin Neurol 2011;24(5):475-83.
- Saporta ASD, Sottile SL, Miller LJ et al. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011;69:22-33.
- Szigeti K, Lupski JR. Charcot-Marie-Tooth disease. Eur J Hum Genet 2009;17:703-10.
- Foley C, Schofield I, Eglon G, et al. Charcot-Marie-Tooth disease in Northern England. J Neurol Neurosur Ps [Epub ahead of print]. 54
- Braathen GJ, Sand JC, Lobato A, et al. Genetic epidemiology of Charcot–Marie–Tooth in the general population. Eur J Neurol 2010;18(1):39-48.
- Reilly MM. Classification and diagnosis of the inherited neuropathies. Ann Indian Acad Neurol 2009;12(2):80-8.
- Reilly MM, Shy ME. Diagnostic and new treatments in genetic neuropathies. J Neurol Neurosur Ps 2009;80:1304-14.
- Siskind CE, Shy ME. Genetics of neuropathies. Semin Neurol 2011;31(5):494-505.
- Fledrich R, Schlotter-Weigel B, Schnizer TJ, et al. A rat model of Charcot-Marie-Tooth disease 1A recapitulates disease variability and supplies biomarkers of axonal loss in patients. Brain 2012;135(Pt1):72–87.
- Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genom Hum G 2008;9:387-402.
- Voelkerding KV, Dames S, Durtschi JD. Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy. J Mol Diagn 2010;12(5):539-551.
- Lupski JR, Reid JG,Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. New Engl J Med 2010,362:1181-1191.
- Weedon MN, Hastings R, Caswell R et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 2011;89:308-12.
- Montenegro G, Powell E, Huang J, et al. Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth family. Ann Neurol 2011;69:464-470.
- Singleton AB. Exome sequencing: a transformative technology. Lancet Neurol 2011;10:942-6.
- Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian gene discovery. Nat Rev Genet 2011;12:745-55.
- Berciano J. Molecular diagnosis of Charcot-Marie-Tooth disease. Nat Rev Neurol 2011;7(6):305-6.
- Patzkó E, Shy ME. Update on Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep 2011;11:78-88.
- MacArthur DG, Balasubramanian S, Frankish A, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012;335:823-8.
- Züchner S. Whole genome sequencing identifies causal variants in CMT. Nat Rev Neurol 2010;6:424-5.
- Kochanski A. How to assess the pathogenicity of mutations in Charcot-Marie-Tooth disease and other diseases? J Appl Genet 2006;47(3):255-60.
- Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet 2011;12:628-40.