


Summary
- ARSACS is a rare autosomal recessive disorder characterised clinically by cerebellar ataxia, spasticity, pyramidal signs, peripheral neuropathy and skeletal foot abnormalities
- It is caused by mutations in the SACS gene which encodes the 520kDa protein sacsin
- The availability of the genetic test has extended the clinical spectrum to include examples without spasticity or ataxia
- Retinal examination shows retinal striations on fundoscopy and thickening of the retinal nerve fibre layer on ocular coherence tomography (OCT) in the majority of the cases
- Neurophysiological studies show an early demyelinating sensorimotor neuropathy with progressive axonal degeneration
- MR imaging shows superior vermian cerebellar atrophy, thinning of the cervical spinal cord and pontine linear hypointensities
ARSACS is a rare and disabling, slowly progressive neurodegenerative disorder characterised by cerebellar ataxia, spasticity, pyramidal signs, peripheral neuropathy, skeletal foot abnormalities and thickening of the retinal nerve fibre layer (RNFL) visible on fundoscopy and by ocular coherence tomography (OCT). The condition was first considered to be confined at relatively high frequency to the descendants of founder populations in the Charlevoix and Saguenay-Lac Saint Jean regions of North-Eastern Québec, but the discovery of the causative SACS gene has permitted its identification throughout the world and has extended the diversity of mutations known, and the spectrum of clinical features described. ARSACS is now recognised as one of the important causes of autosomal recessive ataxia. In this review, we summarise the clinical, genetic and pathophysiological features of this condition, and the investigations used in its diagnosis.
History and origins
Québec was one of the first regions of North America to be colonised by Europeans and the majority of French Canadians living in Québec Province today are thought to descend from these original founders. As a result, a number of rare neurogenetic disorders show increased prevalence or local variants in this region, including Friedreich’s ataxia (FRDA), and other hereditary ataxias, spastic parapareses and neuropathies.1 Québec City was founded in 1608 under the rule of the French crown and between 1665 and 1725, around forty families migrated from there to the isolated mountainous region of Charlevoix on the north shore of the Saint Lawrence River. Between 1838 and 1855, further families moved from here to the more distant Saguenay and Lac Saint Jean regions to the north. It is estimated that the carrier frequency of SACS mutations is 1/22 in these regions.
The clinical syndrome of ARSACS was first described in 1978 in these populations,2 and this community of more than 300 affected individuals remains the most numerous and the most extensively studied. The clinical phenotype was remarkably homogeneous, probably because more than 92% of individuals shared the same mutation, and more than 96% shared one of two mutations.3 The causative gene was first described in 20004 enabling the subsequent identification of cases in Europe, North Africa, Turkey, Japan and Brazil5 with considerable phenotypic heterogeneity, so that now neither spasticity nor ataxia must be regarded as an obligate feature of the condition.6
Genetics and sacsin protein function
The causative gene on chromosome 13q12.12 is named SACS and was originally thought to contain a single giant exon4 (see Figure 1). A further 8 coding exons and a tenth non-coding exon have subsequently been identified upstream of this, forming a 13,737bp open reading frame.7 More than 100 different pathogenic mutations have now been described,3, 8 largely missense, nonsense, frameshift and splice-site mutations spread over 6 of the 10 exons, but still primarily in the giant exon 10.3 Large deletions have also been described causing atypical features such as late onset or prominent hearing loss. These have included an intragenic deletion of exons 3-5,8 deletion of the whole gene9 and deletion of SACS and the contiguous gene SGCG causing concomitant limb girdle muscular dystrophy type 2c.10
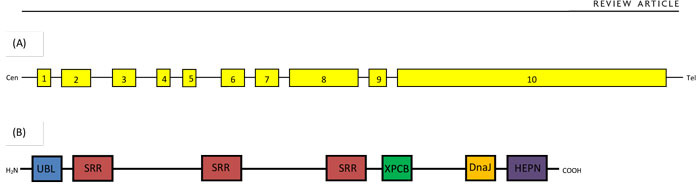
The gene encodes a 4,579 amino acid 520kDa protein called sacsin (see Figure 1) whose true function is currently not fully understood11 but may have a protective effect against mutant ataxin-1.12 Sacsin is most highly expressed in cerebellar Purkinje cells; thalamic, midbrain, precerebellar and brainstem nuclei; and large pyramidal forebrain neurones.12 Within cells, it is localised to the cytoplasm and mitochondria, and may have a role in the regulation of mitochondrial dynamics, leading to mitochondrial mislocalisation and dysfunction.11
Histopathology
Nerve biopsies most consistently show a marked decrement in large myelinated fibres. More variably, axonal degeneration with condensation of the axoplasm, increased collagen pockets and accumulation of mitochondria and vesicular bodies is seen, sometimes with regenerative axonal sprouting. Thinning of the myelin sheaths with rare onion bulbs may also be observed.13-15 Taken together, these findings suggest an axonal neuropathy associated with some demyelinating features. Muscle biopsies are typical of neurogenic atrophy.
Two post-mortem examinations of patients with ARSACS have been published.16 The first in a young patient, showed a grossly atrophied superior cerebellar vermis especially in the anterior structures (central lobule and culmen). No changes were seen in the dentate nucleus and inferior olives. The molecular and granular cell layers were thinned with practically absent Purkinje cells. The pyramids, lateral and anterior corticospinal tracts and posterior spinocerebellar tracts all showed significant loss of myelin staining, particularly the lateral corticospinal tracts. The corticospinal changes were more marked in the upper cord, whereas the spinocerebellar changes were more marked caudally. The second, in an older patient, showed similar findings although to a more pronounced degree. Swollen thalamic and cerebellar cortical neurones were seen, suggestive of a storage disorder. Most of these cells showed dense, lipofuscin-like granules within lysosomes, although testing of an extensive panel of lysosomal enzymes was normal. Interestingly, lipofuscin deposits have also been seen in the skin biopsy of a patient with ARSACS performed to exclude Lafora body disease.17 Peripheral nerve and muscle biopsies have not shown lipofuscin deposits. The significance of this finding therefore remains unclear.
Clinical aspects
Much of the clinical knowledge of ARSACS is based on the relatively homogeneous Québecois cases. However, subsequently identified cases from elsewhere have demonstrated a genetic and clinical variability which continues to extend the phenotypic description of this condition. In the Québecois cases, unsteadiness was noted from beginning to walk (12-18 months old) which was rarely delayed.16,18 80% initially presented because of walking difficulties and a tendency to fall. At first presentation, approximately 60% were found to have limb ataxia, 80% showed some pyramidal involvement and 50% had both pyramidal and cerebellar involvement. There was no clinical evidence of neuropathy at presentation in the form of pes cavus or intrinsic hand muscle wasting.19 There is some evidence that age at onset may be a little later in non-Québecois cases, particularly in Japanese and Tunisian cases.20 In a series of 17 Belgian patients, 29% had onset at or after age 20 with one as late as 40.8 There is no male-female preponderance.
Thus, limb and gait ataxia are early signs followed by spasticity, which is more prominent in the lower limbs. Spasticity and ataxia affect speech, which is often slightly slurred in childhood and can become explosive in adulthood. Dysphagia is usually mild or absent.18,21 Plantar reactions are frequently upgoing from childhood. Eye movements show horizontal bidirectional nystagmus, saccadic alterations of smooth ocular pursuit and saccadic dysmetria.18 Supranuclear gaze palsy has been described in one case.17 In the Québecois cases, by the age of 10 more than 90% showed both pyramidal and cerebellar involvement.19 Muscle cramps may be a troublesome feature. Progression of symptoms is slow. In the Québecois cases, only 4% used a wheelchair before the age of 18.19 Mean age to being wheelchair-bound was around 40 (range 17-58) and to death around 50 (range 21-72).18
From childhood, deep tendon reflexes are frequently increased, but by adulthood, may diminish or become absent due to progressive neuropathy. Ankle jerks are commonly absent whilst knee jerks may be hyperreflexic but patients may have a very mixed and asymmetric picture. Sensory deficits usually appear later and progressively into adulthood, involving vibrational sense more than proprioception and cutaneous sensation. Distal amyotrophy also appears progressively later in the condition.18 The combination of early spasticity and progressive neuropathy commonly causes skeletal abnormalities of the foot including pes cavus, talipes equinus or varus, and hammer or clawed toes. Unlike FRDA, spinal scoliosis is not a prominent feature18 but has been described in Tunisian14 and Italian21 series. Straight dorsal spine has been described in a Spanish series.22 In the hands, swan-neck deformity of the fingers and claw hands have been described2,15 with dystonia sometimes causing abnormal posturing of the hands and neck.23
Cognition is generally preserved particularly on tests of verbal function, but visuospatial handling may be diminished and deteriorate with time.16 Cognitive impairment may be a more prominent feature amongst non-Québecois patients, with intellectual impairment and dementia described in patients from Japan, Italy and Turkey.15 Although cerebellar eye signs may cause visual disruption, optic nerve and retinal function are not generally affected with normal acuity, fields and colour vision, despite the presence of thickened retinal nerve fibres (see below).18 Hearing loss is not generally found but may be more prominent amongst cases involving SACS gene deletions.9, 21, 24
Bladder and bowel symptoms are not well-studied in ARSACS although urinary urge incontinence has been most commonly described.2,6,18,21,23 Faecal incontinence and constipation may also be a problem in patients with long disease duration.18 Co-existent epilepsy has been described in a minority of cases and it remains unclear whether this is a definite association.17,23 It appears more common in the Québecois cases, occurring in more than 15% in one series.19 Frequent abnormal electroencephalographic features have also been described (see below).
Currently no clinical diagnostic criteria exist for ARSACS. The descriptive clinical features published by Bouchard, et al16,18 have come closest to this, although may be more representative of the Québecois cases.
Clinical features of ARSACS
(modified from Bouchard, et al16,18)
Onset
- Unsteadiness at gait initiation
Progressive signs
- Mostly spastic ataxia of the four limbs
- Slurred and dysrythmic speech
- Discrete to severe distal amyotrophy
- Absent ankle jerks after 25 years of age
Early non-progressive signs
- Increased deep tendon reflexes
- Bilateral abnormal plantar response
- Marked saccadic alteration of ocular pursuit
- At funduscopy: prominent retinal nerve fibres radiating from the disc and embedding retinal vessels
Positive diagnostic tests
- CT or MRI: atrophy of the superior vermis; progressive atrophy of the cerebellar hemispheres and of the cervical spinal cord.
- NCS: axonal neuropathy with absent sensory action potentials and low motor conduction velocities.
Imaging
The predominant radiological manifestations of ARSACS on MRI and CT are marked atrophy of the superior cerebellar vermis with consequent enlargement of the supravermian cisterns and cisterna magna8,15,21,22 (see Figure 2). Posterior fossa arachnoid cysts are also sometimes reported.6 While such prominent cerebellar atrophy is uncommon in FRDA, these findings are also seen in other causes of spinocerebellar ataxia (SCA). More specific appear to be the paramedian, bilaterally symmetrical, parallel, linear hypointensities in the pons on T2 and T2-FLAIR MRI sequences22,25 which some have called ‘pontine tigroid hypointensities’.26 Associated with these may be bilateral T2-FLAIR hyperintensities of the lateral pons at the level of the middle cerebellar peduncles (MCPs).6 The hypo- and hyper-intensities may extend into the MCPs.27 The pons generally may be bulky21 and the MCPs thickened.6,21,22 The pontine striations have not been reported in other causes of ataxia or spastic paraparesis, making them useful in distinguishing ARSACS from these conditions when present. Diffusion tensor imaging (DTI) has shed some light on the underlying nature of these changes and the cause of symptoms in ARSACS, with hyperplastic pontocerebellar fibres at the same level as thin and abnormally placed pyramidal tracts, suggesting that the former may be compressing the latter.21,22
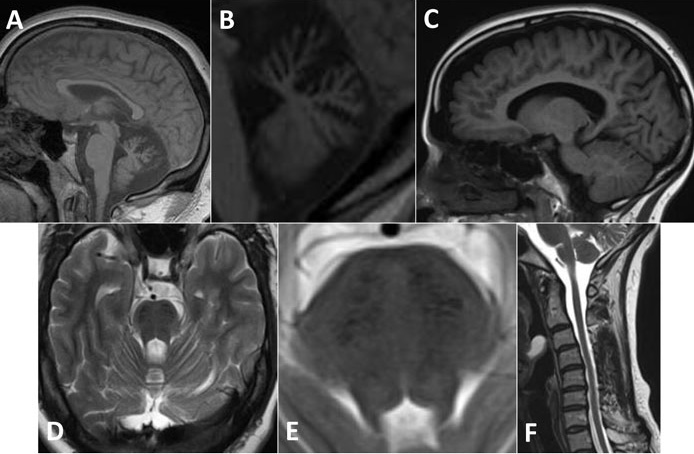
Atrophy of the superior cerebellar peduncles (SCPs), medulla, cervical and thoracic cords has also been observed,18,21 although again not consistently particularly in non-Québecois cases.27 More widespread cerebral atrophy, particularly bilaterally in the parietal lobes,6 may be seen later in the course of the condition but is not as prominent as the cerebellar or cervical atrophy.8,16 Thinning of the corpus callosum and a rim of T2 hyperintensity around the thalami have also variably been reported.6,21 No white matter abnormalities have been seen in either brain or spine,15,18 except in one atypical case in which the explanation was felt to be concomitant multiple sclerosis.26 Single photon emission computed tomography (SPECT) has shown decreased blood flow in the superior cerebellar vermis.28
Thus the salient imaging features of ARSACS are prominent early superior vermian cerebellar atrophy, thinning of the predominantly cervical spinal cord and pontine linear hypointensities.
Neurophysiological studies
Nerve conduction studies show increased distal motor latency and decreased conduction velocities which are more pronounced in the lower limbs than the upper limbs. Typical median nerve conduction velocities are 29-44ms-1 and peroneal nerve 17-35ms-1. This appears to distinguish ARSACS from FRDA in which motor conduction velocities are usually preserved. Motor conduction slowing appears early in life with progressive degeneration which may make compound motor action potentials impossible to detect at the feet by middle age. Sensory nerve conduction is usually of low amplitude or unrecordable, especially in the lower limbs.13,14,18,22 Electromyography shows fibrillations, occasional fasciculations, increased polyphasic action potentials and decreased or absent recruitment, indicating chronic denervation of distal muscles early in the disease process.18,22 Sympathetic skin responses are normal. Somatosensory evoked potentials show a dispersed and delayed cortical response indicating slowed central sensory conduction. Brainstem and visual evoked potentials show increased latencies even in the absence of auditory or visual symptoms.18,22 Electroretinography is normal.21,29 Transcranial magnetic stimulation also shows marked delay in the central pathways.18 Thus, neurophysiological studies suggest an early demyelinating sensorimotor neuropathy with progressive axonal degeneration, and involvement of the central sensory and motor pathways.
Electronystagmography most commonly shows horizontal gaze-evoked nystagmus and impairment of smooth ocular pursuit. There is also impairment of optokinetic nystagmus and defective fixation suppression of caloric nystagmus. Saccades are dysmetric but saccadic velocities are normal.18
Electroencephalography (EEG) reveals abnormalities in 40-60% of patients although frank epilepsy is much less commonly reported.18,21 These abnormalities are non-specific findings indicating involvement of cortical and subcortical structures similar to those reported in FRDA.
Retinal changes
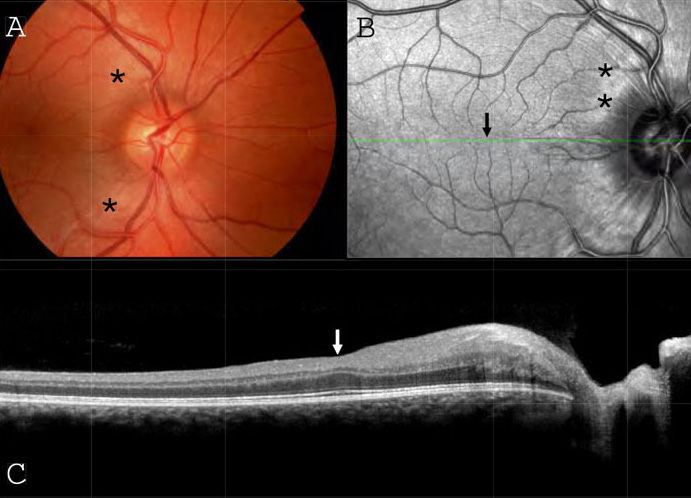
Thickening of the retinal nerve fibre layer (RNFL) is the characteristic retinal change visible on funduscopy in ARSACS (see Figure 3). This appears as prominent streaks emanating in all directions from the optic disc, most striking in the papillomacular bundle and nasal to the disc (where such striations are rarely visible in healthy people). The retinal vessels, which normally lie on the surface of the nerve fibre layer, may be ‘buried’ within this thickened layer obscuring their normally crisp margins.2 These retinal striations are often described in the literature as composed of myelinated18 or hypermyelinated15,21 retinal fibres, although their exact nature remains unknown as no histopathological studies of the eyes in ARSACS have yet been published. Indeed, it is likely that ‘myelinated fibres’ is a misnomer; myelin in the retina is rare, but when present appears opaque and is associated with a corresponding area of visual field loss, whereas the retinal striations in ARSACS are translucent and not associated with any loss of sight.29-33
However, these retinal changes are not consistently observed on funduscopy, particularly in non-Québecois cases of ARSACS. A more sensitive method of detecting them appears to be ocular coherence tomography (OCT). This technique and its uses in neurological disease have previously been reviewed in this journal.34 In ARSACS, OCT shows thickening of the RNFL in all sectors around the disc, with average peripapillary thicknesses of between 119 and 220μm.22,29-33 In the macula, RNFL thickening extends over the fovea and can obscure the foveal pit.31 RNFL thickening is not seen in the context of any other chronic progressive neurodegenerative diseases, only in cases of optic disc swelling associated with intracranial hypertension, optic neuritis or other local pathologies affecting the optic nerve head. Some cases of retinitis pigmentosa have shown thickening of the RNFL although with heavily disrupted outer retinal layers. It is therefore important to interpret the OCT results alongside the clinical history and fundoscopic findings.
Differential diagnosis
FRDA is the commonest cause of autosomal recessive cerebellar ataxia and the chief condition in the differential diagnosis of ARSACS. Retained or brisk reflexes and spasticity are rarely features of FRDA except in atypical late-onset cases known as Friedreich’s ataxia with retained reflexes (FARR).35 Cerebellar atrophy is more pronounced in ARSACS. A striking feature which distinguishes ARSACS from FRDA and other mitochondrial disorders, is the absence of extraneurological features such as diabetes, cardiomyopathy and scoliosis. The electrocardiogram in ARSACS is typically normal, as compared to the frequent existence of repolarisation abnormalities in FRDA. Although mitral valve prolapse was described in the original cases of ARSACS,2 this finding has not been replicated in subsequent studies of families outside Québec.
Ataxia with oculomotor apraxia (AOA) may be distinguished from ARSACS because of the presence of oculomotor apraxia, dystonia, chorea and the absence of pyramidal features. AOA type 1 is associated with low levels of serum albumin and elevated levels of low density lipoproteins (LDLs), whilst AOA type 2 shows elevated levels of α-fetoprotein (AFP). Ataxia telangiectasia has many features in common with AOA together with cutaneous and scleral telangiectasiae, diabetes, immunodeficiency and sensitivity to radiation causing tumours.19
Late-onset Alexander’s disease may have onset in adolescence and have a presentation similar to ARSACS. Cerebellar atrophy is less prominent and there may be periventricular white matter changes on MRI which are not seen in ARSACS. Cerebrotendinous xanthomatosis has onset in infancy but is often associated with diarrhoea, cataracts and tendon xanthomata, and is identifiable because of elevated serum cholestanol and urinary bile alcohols. Of the hereditary spastic parapareses (HSPs), HSP7 may be one of the most common to be complicated by ataxia, although onset is generally in adulthood. HSP 11, 15, 20, 21 and 27 may also present with ataxia, although often show distinguishing features.36,37 In the spinocerebellar ataxias (SCAs), cerebellar ataxia generally predominates and inheritance is autosomal dominant. Amongst these, SCA1 and SCA3 (Machado-Joseph disease) are the most common which can present with a spastic paraparesis, however in both of these conditions, age at onset is in adulthood.38,39
If neuropathy dominates the clinical picture over spasticity and ataxia, Charcot-Marie-Tooth (CMT) disease may be considered.6 A number of other rare causes of ataxia or spastic paraparesis may need to be considered including spastic ataxia types 1 to 5 (SPAX1-5), abetalipoproteinaemia, ataxia with vitamin E deficiency (AVED), ataxia with coenzyme Q10 deficiency, Niemann-Pick disease type C, Refsum’s disease and autosomal recessive cerebellar ataxia type 2 (ARCA2), for which genetic or metabolic tests are available.40
Once acquired causes of spastic ataxia have been excluded, the combination of age at onset, suspected mode of inheritance, associated clinical, neuroimaging, neurophysiological and other features should guide genetic testing. In the future, next generation41 and whole exome sequencing will allow parallel testing of multiple suspected genes, although it will remain vital to interpret the results in terms of pre-existing suspicions from careful clinical phenotyping.
Conclusions
The triad of early-onset ataxia, spasticity and axonal-demyelinating neuropathy, together with sporadic or autosomal recessive inheritance, prominent superior cerebellar and cervical atrophy on MRI and no extraneurological features, should provoke the suspicion of ARSACS. Many formes frustes will continue to be described as genetic techniques permit the identification of more cases. The presence of pontine linear hypointensities on MRI and thickened retinal nerve fibres on OCT, appear to be sensitive markers of ARSACS. All suspected cases should therefore undergo these two tests. Cellular and animal models, and molecular biological techniques are beginning to elucidate the underlying pathophysiology of this condition which may permit the first interventional trials.
References
- Dupré N, Bouchard JP, Brais B, Rouleau GA. Hereditary ataxia, spastic paraparesis and neuropathy in the French-Canadian population. Can J Neurol Sci 2006;33(2):149-57.
- Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci 1978;5(1):61-9.
- Thiffault I, Dicaire MJ, Tetreault M, et al. Diversity of ARSACS mutations in French-Canadians. Can J Neurol Sci 2013;40(1):61-6.
- Engert JC, Berube P, Mercier J, et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Gen 2000;24(2):120-5.
- Bouhlal Y, Amouri R, El Euch-Fayeche G, Hentati F. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: an overview. Parkinsonism Rel Dis 2011;17(6):418-22.
- Synofzik M, Soehn AS, Gburek-Augustat J, et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis 2013;8:41.
- Ouyang Y, Takiyama Y, Sakoe K, et al. Sacsin-related ataxia (ARSACS): expanding the genotype upstream from the gigantic exon. Neurol 2006;66(7):1103-4.
- Baets J, Deconinck T, Smets K, et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurol 2010;75(13):1181-8.
- Breckpot J, Takiyama Y, Thienpont B, et al. A novel genomic disorder: a deletion of the SACS gene leading to spastic ataxia of Charlevoix-Saguenay. Eur J Hum Gen 2008;16(9):1050-4.
- McMillan HJ, Carter MT, Jacob PJ, et al. Homozygous contiguous gene deletion of 13q12 causing LGMD2C and ARSACS in the same patient. Muscle Nerve 2009;39(3):396-9.
- Girard M, Lariviere R, Parfitt DA, et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc Nat Acad Sci USA 2012;109(5):1661-6.
- Parfitt DA, Michael GJ, Vermeulen EG, et al. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum Mol Gen 2009;18(9):1556-65.
- Peyronnard JM, Charron L, Barbeau A. The Neuropathology of Charlevoix-Saguenay Ataxia: An Electrophysiological and Pathological Study. Can J Neurol Sci 1979;6(2):199-203.
- El Euch-Fayache G, Lalani I, Amouri R, et al. Phenotypic features and genetic findings in sacsin-related autosomal recessive ataxia in Tunisia. Arch Neurol 2003;60(7):982-8.
- Takiyama Y. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuropath 2006;26(4):368-75.
- Bouchard JP, Richter A, Mathieu J, et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuromusc Dis 1998;8(7):474-9.
- Stevens JC, Murphy SM, Davagnanam I, et al. The ARSACS phenotype can include supranuclear gaze palsy and skin lipofuscin deposits. J Neurol Neurosurg Psychiatry 2013;84(1):114-6.
- Bouchard JP. Recessive spastic ataxia of Charlevoix-Saguenay. Handbook Clin Neurol 1991;16(60):451-9.
- Duquette A, Brais B, Bouchard JP, Mathieu J. Clinical presentation and early evolution of spastic ataxia of Charlevoix-Saguenay. Mov Dis 2013 Aug 2. doi: 10.1002/mds.25604. [Epub ahead of print]
- Takiyama Y. Sacsinopathies: sacsin-related ataxia. Cerebellum 2007;6(4):353-9.
- Prodi E, Grisoli M, Panzeri M, et al. Supratentorial and pontine MRI abnormalities characterize recessive spastic ataxia of Charlevoix-Saguenay. A comprehensive study of an Italian series. Eur J Neurol 2013;20(1):138-46.
- Gazulla J, Benavente I, Vela AC, et al. New findings in the ataxia of Charlevoix-Saguenay. Journal of neurology 2012;259(5):869-78.
- Vermeer S, Meijer RP, Pijl BJ, et al. ARSACS in the Dutch population: a frequent cause of early-onset cerebellar ataxia. Neurogenetics 2008;9(3):207-14.
- Terracciano A, Casali C, Grieco GS, et al. An inherited large-scale rearrangement in SACS associated with spastic ataxia and hearing loss. Neurogenetics 2009;10(2):151-5.
- Martin MH, Bouchard JP, Sylvain M, St-Onge O, Truchon S. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: a report of MR imaging in 5 patients. Am J Neuroradiol 2007;28(8):1606-8.
- Terracciano A, Foulds NC, Ditchfield A, et al. Pseudodominant inheritance of spastic ataxia of Charlevoix-Saguenay. Neurol 2010;74(14):1152-4.
- Shimazaki H, Takiyama Y, Honda J, et al. Middle cerebellar peduncles and Pontine T2 hypointensities in ARSACS. J Neuroimaging 2013;23(1):82-5.
- Shimazaki H, Sakoe K, Niijima K, Nakano I, Takiyama Y. An unusual case of a spasticity-lacking phenotype with a novel SACS mutation. J Neurol Sci 2007;255(1-2):87-9.
- Desserre J, Devos D, Sautiere BG, et al. Thickening of peripapillar retinal fibers for the diagnosis of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Cerebellum 2011;10(4):758-62.
- Garcia-Martin E, Pablo LE, Gazulla J, et al. Retinal nerve fibre layer thickness in ARSACS: myelination or hypertrophy? Br J Ophthalmol 2013;97(2):238-41.
- Nethisinghe S, Clayton L, Vermeer S, et al. Retinal Imaging in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Neuro-Ophthalmol 2011;35(4):197-201.
- Pablo LE, Garcia-Martin E, Gazulla J, et al. Retinal nerve fiber hypertrophy in ataxia of Charlevoix-Saguenay patients. Mol Vis 2011;17:1871-6.
- Vingolo EM, Di Fabio R, Salvatore S, et al. Myelinated retinal fibers in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Eur J Neurol 2011;18(9):1187-90.
- Raftopoulos R, Trip A. The Application of Optical Coherence Tomography (OCT) in Neurological Disease. ACNR 2012;12(2):30-3.
- Parkinson MH, Boesch S, Nachbauer W, Mariotti C, Giunti P. Clinical features of Friedreich’s ataxia: classical and atypical phenotypes. J Neurochem 2013;126:103-17.
- de Bot ST, Willemsen MAAP, Vermeer S, et al. Reviewing the genetic causes of spastic-ataxias. Neurol 2012;79(14):1507-14.
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol 2008;7(12):1127-38.
- Giunti P, Sweeney MG, Spadaro M, et al. The trinucleotide repeat expansion on chromosome 6p (SCA1) in autosomal dominant cerebellar ataxias. Brain 1994;117(4):645-9.
- Giunti P, Sweeney MG, Harding AE. Detection of the Machado-Joseph disease/spinocerebellar ataxia three trinucleotide repeat expansion in families with autosomal dominant motor disorders, including the Drew family of Walworth. Brain 1995;118(5):1077-85.
- Anheim M, Tranchant C, Koenig M. The Autosomal Recessive Cerebellar Ataxias. New Engl J Med 2012;366(7):636-46.
- Németh AH, Kwasniewska AC, Lise S, et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain 2013;136(10):3106-18.