



Abstract
Craniosynostosis is a group of conditions characterised by the premature fusion of one or more cranial vault sutures. This may lead to abnormal cranial development with severe skull and craniofacial deformities and if the condition is left untreated, other complications such as raised intracranial pressure and cranial growth restriction may be implicated.
Craniosynostosis can arise as part of a genetic syndrome, or nonsyndromically where the pathophysiology remains less clear. Occurring in 1 in 2,000 to 2,500 live births, diagnosis is carried out shortly after birth and treatment of craniosynostosis mostly involves surgery varying from less invasive procedures in those patients diagnosed early to single or repeated open calvarial reconstruction in the more complex cases.
This article reviews the different types of craniosynostosis with their variable presentations, underlying genetic mutations, associated complications and neuro-psychological outcomes before discussing its management with distinct emphasis on surgical treatment options within a multidisciplinary team.
Introduction
Craniosynostosis is a group of conditions characterised by premature fusion of one or more of the cranial vault sutures. This can lead to abnormal cranial development and give rise to severe skull and craniofacial deformities. Craniosynostosis can arise as part of syndromes, with specific gene mutations resulting in other non-cranial manifestations in addition to synostosis, or nonsyndromically where the pathophysiology remains less clear. Both types of craniosynostosis can be familial or sporadic. Occurring in 1 in 2,000 to 2,500 live births,1-3 infants are diagnosed at birth or within a few months thereafter4 and should preferably have treatment within their first year of life.5 If the condition is left untreated, craniosynostosis can lead to further deformity and other complications such as raised intracranial pressure6-7 and cranial growth restriction. The treatment mostly involves surgery varying from less invasive procedures in those patients diagnosed early8-10 to single or repeated open calvarial reconstruction in the more complex cases.11,12 There are a number of clinicians involved in the care of children with this condition, highlighting the importance of a multidisciplinary team. This article will review the different types of craniosynostoses with their variable presentations, the underlying genetic mutations, complications and neuro-psychological outcomes before discussing its management with distinct emphasis on surgical treatment options within a multidisciplinary team.
Embryology
The human cranium is divided into the neurocranium housing the brain, and the viscerocranium, comprising the face. The neurocranium forms from embryonic mesenchyme of neural crest (frontal bone) and paraxial mesoderm (parietal bone) origin,13 which surrounds the brain and forms primary ossification centres termed bone spicules. Each island of mineralised tissue migrates and undergoes intramembranous ossification to form the plates of the neurocranium. These plates remain separated in early infanthood, allowing for passage during labour and continued growth of the brain after birth. The metopic suture fuses between 3 to 9 months whilst the sagittal, coronal and lambdoid sutures do not stop growing until the second decade and eventually fuse within the third decade.14-16 Each plate approaches one another but remains separated by the formation of a suture: the two halves of the frontal bone by the metopic suture; the frontal and parietal bones by the sagittal suture; the two halves of the parietal bone by the coronal suture; and the parietal and occipital bones by the lambdoid suture. Fontanelles, namely membrane-covered “soft spots”, are located at the intersection of sutures: the larger anterior fontanelle at the intersection of the metopic, coronal and sagittal sutures and the smaller posterior fontanelle at the intersection of the sagittal and the lambdoid sutures. These fontanelles usually fuse by the age of 18 months and 3 to 6 months respectively.14
Types
Premature fusion of the sutures implicates that the normal growth of the neurocranium is arrested at one or more sites. In order to accommodate the growing brain, compensatory growth occurs at other sites leading to abnormal cranial development and deformity. This was described in 1851 through Virchow’s law that states that if a suture prematurely fuses, growth is arrested perpendicular to the suture and is increased parallel to it.17,18 Thus, it explains the characteristic and predictable patterns of cranial growth that occur as a result of the premature fusion of distinctive sutures (see Figure 1, adapted from Senarath-Yapa et al., 201219).

Sagittal synostosis is the most common type, accounting for 40-55% of nonsyndromic craniosynostosis.17,20 Caused by premature fusion of the sagittal suture, growth is arrested in the transverse direction and increased in the anteroposterior direction, resulting in an anteroposterior elongation with frontal bossing and occipital prominence. This characteristic “long boat” shape skull is termed scaphocephaly (derived from skaphos: Greek term for skiff).
Coronal synostosis has been superseded by metopic as the second most common nonsyndromic synostosis as several studies have shown over the past decade.3,21,22 It occurs in 20-24% of nonsyndromic cases23,24 and can be either unilateral or bilateral.
Premature fusion of the coronal suture bilaterally produces the opposite pattern of abnormal growth to sagittal synostosis, arresting growth in the anteroposterior direction and increased growth in the transverse direction, producing a short wide head called brachycephaly (from the Greek term brachkus for short). Unilateral coronal synostosis causes flattening of the ipsilateral forehead and displacement of the ipsilateral lesser wing of the sphenoid bone superolaterally called the “harlequin eye deformity” since radiographically it has the appearance of a masquerade mask. Other features include ipsilateral nasal deviation and contralateral displacement of the anterior fontanelle. This skull pattern produced by unilateral coronal synostosis is termed anterior plagiocephaly (plagos: Greek for slant).
Metopic synostosis is found in 20-29% of non-syndromic cases but studies have shown increasing prevalence.24,25 Premature fusion of the metopic suture causes arrested growth of the cranium in the transverse direction anteriorly and increased anteroposterior growth. This narrowing of the frontal bone produces a pointed triangular forehead with orbital hypotelorism and a ridge along the fused metopic suture and there may be compensatory posterior growth causing widening of the parietal regions. This is called trigonocephaly (trigonos: Greek term for triangle). However, it is important to note that ridging not infrequently occurs with normal fusion during the first few months of life and does not require surgery.26
Lambdoid synostosis is rare, occurring in 0-5% of non-syndromic cases17,20 and is usually unilateral. Due to premature fusion of one of the lambda sutures there is arrested growth of the ipsilateral occipital region causing ipsilateral occipital flattening, posteroinferior displacement of the ipsilateral ear and tilting of the skull base towards the affected suture. Compensatory growth occurs at the contralateral occipital and frontal regions resulting in contralateral forehead and occipital protuberances as well as inferior mastoid elongation. This posterior slanting shape is called posterior plagiocephaly. Bilateral lambdoid synostosis is very rare and causes symmetrical flattening of the occiput with compensatory heightening the skull. This is called posterior brachycephaly and in combination with posterior sagittal synostosis also known as the “Mercedes Benz” sign due to the changes on the X-rays.27 Bilateral lambdoid synostosis is associated with a Chiari I abnormality (with protrusion of cerebellar tonsils through the Foramen magnum) and can appear similar to brachycephaly due to coronal synostosis.
A similar presentation, and by far the most common one, can occur in positional plagiocephaly (“moulding”), a prevalent acquired cranial asymmetry that emerges at 6 weeks of age and can largely be attributed to the supine sleeping position recommended for infant safety (in the UK generally referred to as the “Back to Sleep” campaign for the prevention of Sudden Infant Death Syndrome).28-30
The two can be difficult to distinguish (see Figure 2), but the ipsilateral ear is anteriorly displaced in positional plagiocephaly and skull base tilt is absent. Positional plagiocephaly is asserted to be benign and may resolve spontaneously in some cases32,33 or with simple measures such as position changes, “tummy time” and physical therapy for any torticollis that may be present.30,31

Although orthotic (“moulding”) helmets are frequently used (particularly in Europe and the USA),33,34 Wijk et al. demonstrated in HEADS (HElmet therapy Assessment in Deformed Skulls), a single blinded, randomised controlled trial, that there is no benefit from the administration of a moulding helmet and discouraged its use due to the association with large costs and prevalent side effects.35 The overall consensus in the UK is to not recommend it.
Synostoses occur in multiple sutures in 5-15% of non-syndromic cases,17,20 presenting with more complex deformities. Synostosis of three or more sutures is referred to as pansynostosis36,37 and can present either with microcephaly or as a “Kleeblattschädel” (cloverleaf skull), named due to the bulging of the frontal and temporal bones giving rise to a tri-lobular shaped skull.
Genetics
Nonsyndromic craniosynostosis accounts for approximately 85% of cases3 and although positive family histories have been reported,38,39 the aetiology remains unknown. However, one cohort study genetic analysis found single gene mutations in FGFR2, FGFR3, TWIST1 and EFNB1 in 11 out of 204 (5.4%) of non-syndromic cases, 9/11 of which were unilateral or bilateral coronal.40
Other factors, including increased thyroid hormone level during pregnancy, and environmental stimuli such as head compression in utero, maternal smoking and teratogenic medications have also been implicated.41 Of particular note is the association between maternal use of sodium valproate and metopic craniosynostosis.42 On the other hand, most types of syndromic craniosynostoses are inherited in an autosomal dominant fashion43,44 and genetic analysis studies have provided strong links to a number of genes.43
One such group of genes implicated is the fibroblast growth factor receptor family, of which mutations in genes encoding FGFR1, FGFR2 and FGFR3 have been found in syndromic craniosynostoses. These are receptor tyrosine kinases that undergo auto-phosphorylation upon fibroblast growth factor binding and are involved in a vast range of cell functions and developmental processes.45,46 Indeed, targeted mutagenesis of individual FGFR isotypes has been shown to lead to both lethal or viable defects in embryological development such as gastrulation,45 placenta and limb bud formation,47,48 organogenesis49 and bone ossification.50
FGFR mutation results in gain of function causing abundant activation of the FGF/FGFR signalling pathway, which is then leading to expression of runt-related transcription-factor 2 (RUNX2). The result is early onset differentiation of mesenchyme cells into osteoblasts that deposit bone and eventually lead to premature suture closure.51,52 FGFR1 mutations have been identified in Pfeiffer and Jackson Weiss syndromes; FGFR2 mutations in Crouzon, Jackson Weiss, Apert, Pfeiffer and Beare Stevenson syndrome; and FGFR3 mutations in Crouzon syndrome with Acanthosis, Muenke syndrome and Thantophoric dysplasia (see Table 1 and Figure 3). The mechanism resulting in significantly differing phenotypes arising from the same mutation is yet to be fully understood.
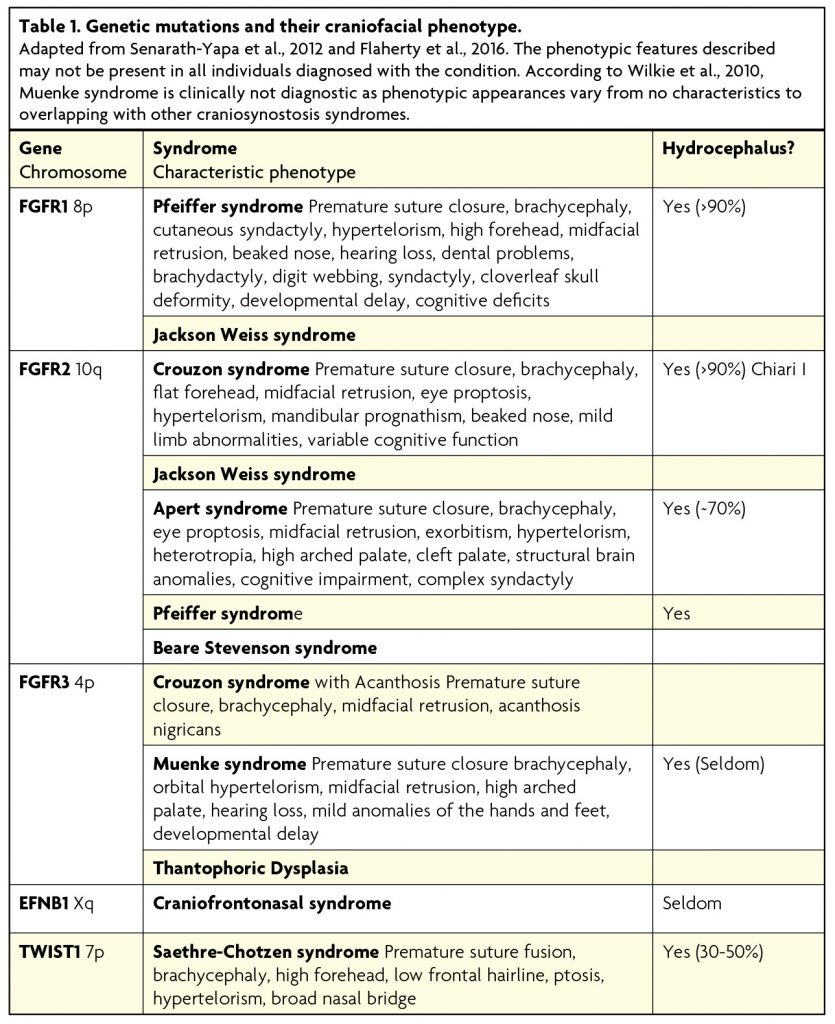
Figure 3. Most common craniosynostosis syndromes
Crouzon syndrome,57 first described by Octave Crouzon in 1935, is the most common of the craniosynostosis syndromes, occurring in 1 in 25,000 live births. Like the majority of the syndromes including Apert, Pfeiffer and Saethre-Chotzen, it follows an autosomal dominant inheritance pattern7 and mutations have been found in FGFR2 and FGFR3. Most commonly affected are the bilateral coronal sutures causing brachycephaly. Also seen is hypertelorism, shallow orbits resulting in exophthalmos, maxillary hypoplasia causing mandibular prognathism, high arched palate and low set ears associated with hearing impairment. Crouzon syndrome is also thought to convey an increased risk of raised intracranial pressure58 and this has been proposed to be due to the early closure of the sagittal and lambdoid sutures.59 As a result cognitive function in individuals with Crouzon syndrome is variable. Additionally, this syndrome may well be progressive in the first 2-3 years of life and even within the same family can have quite marked differences in phenotype.
Apert syndrome is the second most common, found in 1 in 100,000 newborns, the majority of which are sporadic mutations in FGFR2. It also affects the coronal sutures bilaterally causing a brachycephaly60 with hypertelorism, shallow orbits, exophthalmos and high arched palate. However, maxillary hypoplasia is more severe than observed in Crouzon syndrome and can lead to life-threatening airway compromise. Also seen is an anterior open bite, downslanting palpebral fissures, a “parrot beak” nose and syndactyly of the second, third and fourth digits.
Pfeiffer syndrome also occurs in 1 in 100,000 live births, most commonly due to FGFR2 mutations, but FGFR1 mutations have been found in 5% of cases, causing a less severe presentation.61 The coronal, lambdoid and sagittal sutures are all affected, but heterogeneity of the syndrome has led to a classification into three clinical types. Type I is the classic, most common and least severe type associated with turribrachycephaly, hypertelorism, strabismus, maxillary hypoplasia causing mandibular prognathism and characteristic broad thumbs. Type II is more severe, with a cloverleaf skull, severe exophthalmos, hydrocephalus and poor prognosis. Type III is very similar to type II but lacks the cloverleaf skull.62
Saethre-Chotzen is found in 1 in 25,000 to 50,000 newborns and caused by mutations in TWIST1. The phenotype is heterogenous and synostosis can be bicoronal, unicoronal, sagittal, metopic or multisutural63 leading to a great variety of head shapes. Other features include a low hairline, ptosis, facial asymmetry and ear deformities. Additionally, syndactyly of the second and third digits may be present. Overall, Saethre-Chotzen syndrome perhaps displays the widest phenotype of the common syndromic conditions and family members may remain undiagnosed due to portraying mild phenotypic features (e.g. subtle ptosis).
TWIST1 (twist-related protein 1) is another gene linked to craniosynostosis syndromes and mutations have been found in the Saethre-Chotzen syndrome. TWIST1 is a basic loop-helix-loop transcription factor and thought to be involved in determining the lineage of osteoblasts. Cells over-expressing TWIST1 showed decreased response to FGF and remained undifferentiated while cells underexpressing TWIST1 differentiated into a mature osteoblast-like state.53
Therefore, it has been hypothesised that TWIST1 is involved with delaying suture fusion, upstream of FGF. Indeed, the majority of TWIST1 mutations found in Saethre-Chotzen syndrome confer a loss of function through haplo-insufficiency.54 Furthermore, FGFR2 and FGFR3 mutations have also been found in Saethre-Chotzen syndrome55 further supporting a common molecular pathway.
More recently, Zhao et al., 2015 discovered that Gli1+ cells in the suture mesenchyme form the osteogenic front, periosteum, dura and all craniofacial bones, and are involved in injury repair.56 Ablation of Gli1+ cells in mice was found to cause pansynostosis, arresting of skull growth and reduced injury repair. Moreover, the Gli1+ population was reduced in Twist1+/–mice, a widely used model of craniosynostosis mimicking the TWIST1 mutation in Saethre-Chotzen syndrome, and causing increased mesenchyme apoptosis and reduced proliferation. Therefore, the authors showed that Gli1+ cells in the suture mesenchyme form the osteogenic stem cells of the craniofacial sutures and that pathogenesis of craniosynostosis may be due to reduced numbers of Gli1+ cells.
Associated complications
Each type of craniosynostosis can vary in its severity of phenotypic features. In particular, sagittal and metopic suture synostosis may show a very mild clinical presentation in which only one bone ridge at the afflicted suture is visible and/or palpable. Therefore, parents are often confronted with health care professionals who do not recognise the craniosynostosis in a timely manner shortly after childbirth. This may not only cause distress for the parents but also lead to delayed diagnosis and treatment.64 In syndromic and complex nonsyndromic craniosynostoses the patients may suffer from cognitive impairment and raised intracranial pressure (ICP). Several syndromic craniosynostoses are associated with skeletal hypoplasia of the midface resulting in a narrowed airway. In approximately 50% of cases this leads to OSAS (obstructive sleep apnoea syndrome). Other risks and complications include cornea injury due to exorbitism, malocclusion and aesthetic/psychosocial problems. Associated intracranial abnormalities in syndromic craniosynostoses are increased ICP, Chiari I malformation, ventriculomegaly and hydrocephalus. Hearing loss is described for all types of syndromic craniosynostoses. Visual pathologies such as astigmatism and strabismus are very frequent in syndromic craniosynostoses. In nonsyndromic craniosynostosis, specifically unicoronal craniosynostosis, children are at risk of developing astigmatism in the eye opposed to the coronal suture synostosis.65 Limb deformities are largely restricted to syndromic craniosynostoses, and notably associated to the Apert syndrome. Both types of craniosynostosis, nonsyndromic and syndromic, may co-occur with cognitive and behavioural impairments. These are either intrinsic due to the congenital defect or secondary to intracranial hypertension or physical deformities. Interestingly, there is continued debate on decreased intracranial volumes, hydrocephalus and raised ICP in patients with single-suture craniosynostosis.27 So far, there is little to no difference in intracranial volumes among various types of craniosynostoses to be found.66,67 Similarly, there was no correlation between hydrocephalus and nonsyndromic craniosynostosis established,68 unless there is bilateral involvement of the lambdoid suture.
However, several studies have shown that children with nonsyndromic craniosynostosis are at high risk of developing intracranial hypertension.58 In fact, elevated intracranial pressure was found in 24-30% of nonsyndromic craniosynostoses.6,69 Yet in 1982, Renier et al.6 reported abnormal ICP recordings (meaning ≥15mmHg during Slow-Wave sleep) in 14% of cases where only one suture is involved and in 47% of cases with multiple sutures intricated. However, in most nonsyndromic cases indication for surgery remains cosmetic. Invasive ICP monitoring is reserved for children with visual and/or developmental deficits, in cases where surgery has been refused and the head circumference is falling off or they have a “Copper beaten skull” on X-ray – although this is a weak clinical sign.
Apart from its association with intracranial hypertension, premature fusion of cranial sutures is also known to affect the underlying brain morphology. In a series of studies, conducted by Aldridge et al. from 2002 to 2005, the authors demonstrated that both cortical and subcortical structures of the central nervous system are dysmorphic in craniosynostosis. Specifically, studies of brain morphology in cases of sagittal and unicoronal synostosis have demonstrated that changes in the brain’s structure are found in adjacent as well as distant and in subcortical regions away from the fused suture.70-72
The highest percentage of associated intra- and extracranial midline problems can be found in children with metopic synostosis. These patients also most commonly present with an IQ deficit. Birth weight, parental age and sodium valproate use during pregnancy have been identified as potential risk factors for the development of metopic craniosynostosis.42,73
Hydrocephalus and tonsillar descent (Chiari I malformation) merit a specific discussion. Chiari I (for the purpose of this article refers to tonsillar descent and crowding of the foramen magnum) has a clear association with the syndromic craniosynostoses shown in Figure 3. An association between non-syndromic lambdoid synostosis (and not other sutures) and Chiari I has also been noted.74 Chiari I and craniosynostosis co-existing have a significant association with syringomyelia,74 which needs to be taken into account when evaluating and imaging these children. One of the hypotheses for the aetiology of Chiari malformation is the “box being too small for the contents” due to occipital hypoplasia. Craniosynostosis, whilst obviously not due to occipital hypoplasia, results in the net same outcome of the skull being disproportionately too small for the brain. This provides a plausible mechanism for the association between Chiari and craniosynostosis as well as potentially giving greater insights into the pathogenesis of Chiari malformation itself.
Hydrocephalus associated with craniosynostosis is common. There is variation in the reported figures for hydrocephalus across the literature, but overall syndromic craniosynostosis is associated with hydrocephalus in up to 30-70% of cases,68,75 as opposed to nonsyndromic craniosynostoses where it occurs in less than 2%. Furthermore, there is no evidence to suggest any causality between the two in most cases of nonsyndromic synostosis.76
The first and key point is to establish whether one is dealing with genuine hydrocephalus or static ventriculomegaly with no increased pressure. This is not always a straightforward task as the synostosis itself may cause raised ICP and the clinical picture is complex, head circumference is not possible to use and radiological signs may be atypical.
The mechanism of hydrocephalus in craniosynostosis is believed to be a mixture of obstructive and absorptive77 arising from venous hypertension.78 Brain atrophy may contribute to static ventriculomegaly, producing a “hydrocephalus ex vacuo” picture.76 Although not the focus of this article, the existence of acquired craniosynostosis secondary to shunt over drainage in the presence of non-fused sutures should be mentioned as well.
- Cranial growth restriction / physical deformity
- Raised ICP
- Cognitive impairment
Multidisciplinary team
With regard to the number of complications that can arise intra- and post-operatively from open cranial vault procedures the multidisciplinary team concept has developed and is widely used. It is largely based around protocols for workup, delivery of anaesthesia, streamlined surgical procedures and complex post-operative care and assessment.79
The involved specialties usually include Plastic Surgery, Neurosurgery, Otolaryngology, Dentistry, Audiology, Ophthalmology, Speech & Language therapy, Developmental Paediatrics, Neuropsychology, Medical Genetics, Social Work and Nursing Care. Other specialists, such as cardiologists and gastroenterologists, may be consulted for management of associated defects and clearance for surgery. Often parents can easily be overwhelmed by all the information discussed when meeting all the different specialists. Moreover, congenital defects involving a child’s face and skull seem to evoke particularly strong emotional responses from the parents, who must contend with a host of potentially stressful events and circumstances, including the infant’s unusual physical appearance, the perspective of potentially life-threatening surgeries ahead, and the possibility of future neuropsychological and educational problems.4
Diagnosis
In order to achieve optimal treatment and satisfactory surgical outcome,80 early diagnosis is essential in children with craniosynostosis. However, patients are not infrequently referred late or not referred at all due to late recognition of the head shape deformity.64
Usually the abnormal skull shape is recognised shortly after birth by either the parents themselves, the treating obstetrician or paediatrician, midwife or general practitioner. The main diagnostic screening tools are physical examination of the skull shape80,81 in combination with taking the history.82,83 The anamnestic flowchart of Bredero may serve as a guideline to distinguish craniosynostosis from positional skull deformities.84 When craniosynostosis is suspected, the paediatrician should refer the child to a craniofacial centre for further diagnostic investigations. X-rays of the skull (A-P, lateral, Towne’s view) are still often performed in cases of suspected craniosynostosis. If the result remains uncertain, the X-ray may be repeated after 1 to 2 months. Alternatively, an experienced investigator can perform ultrasound scanning of the cranial sutures.
CT-scan with 3D-reconstruction is performed as an alternative in some centres.64 Whilst the imaging will also give some detail relating to the brain (hydrocephalus, etc.) it is associated with significantly more radiation and not necessarily of added value in many/most cases of “simple” craniosynostosis.85 Image findings may include bony ridging along the suture, heaping up of bone at the suture, sutural narrowing, and indistinctness of the suture as primary signs of craniosynostosis.86 Secondary signs include an altered calvarial shape, the general changes in shape and timing of closure of fontanels, and other facial anomalies. The lack of growth across a suture commonly results in effacement of the underlying subarachnoid spaces. Patients with craniosynostosis may also have an enlarged subarachnoid space beneath regions of compensatory skull growth.87
In summary, the diagnosis of craniosynostosis is based on the calvarial shape with relation to a calvarial suture. Nonsyndromic craniosynostosis is diagnosed mainly clinically with help of X-rays and CT scans performed in some centres. In contrast to that, syndromic craniosynostosis is often more complex and often requires both CT and MRI imaging to look at the structures within the posterior fossa and venous drainage. For both syndromic and non-syndromic craniosynostosis other investigations should include: regular measurement of the head circumference (and the Cranial Index – width/length), ophthalmology, ENT, neurocognitive, Speech & Language assessments, and where appropriate dental review, measurement of overnight Oxygen saturations (to exclude sleep apnoeas associated with airway problems) and Plastic Surgery opinion for hand and feet abnormalities.
Genetic testing and counselling can assist in making or confirming a specific diagnosis and this may have prognostic implications both for the individual patient but also for future planned pregnancies.64
- Physical examination and history taking
- Diagnostic imaging: X-skull/ ultrasound, 3D-CT scan of the head
- Genetic testing
Neuropsychological outcomes
In syndromic cases surgery is often indicated for morphological (aesthetic) and functional (cognitive, airway, ophthalmic, etc.) reasons. However, in non-syndromic cases, the indication for surgery is still generally considered to be cosmetic. Although, recent evidence suggests that corrective surgery may also positively impact developmental outcomes assessed during long term follow up in non-syndromic synostoses.88
Many of the older studies looking at cognitive outcomes poorly defined mental retardation, lacked control subjects, adequate follow-up periods and valid, standardised psychometric tests. On the other hand, more recent, high quality studies applying the above mentioned principles including formal assessments, such as the Bayley Scales of Infant Development and Wechsler Intelligence Scales for Children, have raised the possibility of mild cognitive impairment even in non-syndromic cases. A systematic review by Knight et al., 201489 of 33 articles with particular emphasis on methodological quality found 10 studies showing developmental delays in motor functioning and cognition, including language, both before and after surgery. Five studies of school-age children with single suture craniosynostosis found Intellectual Quotient to be within the normal range, but three studies found increased learning, behavioural and language deficits documented on medical records or reported by parents, and five studies showed greater speech and language impairment by more formal testing. A few studies uncovered impairment in visual spatial skills, memory and attention, and school performance. Knight at al., 2014 also investigated the literature on correlations between neurodevelopmental outcome and a variety of factors: no articles to date have significantly correlated neurodevelopmental outcomes and brain imaging, severity of deformity, sutures affected, genetics or gender. Interestingly, there is mixed evidence for the association between early surgery and the reduction of neurodevelopmental impairments, with some studies reporting better outcomes with surgery within one year of age and worse outcomes with delayed surgery after four years;90 other studies have reported no such difference.91-93
In addition to cognitive difficulties, psychosocial aspects of craniosynostosis have been investigated. Clearly during early years of infanthood, the major psychosocial burden lies with the parents and this is reflected in the need for parental support. Particularly, parents of a child with syndromic craniosynostosis may have to cope with negative reactions from others, a possible discrepancy between deviating physical appearance and cognition, and be confronted with problems of school choice.64 Once the child grows older and attends school, they may be themselves presented with psychosocial challenges and management of these should in turn focus on the child. A variety of outcomes such as post-traumatic stress, successful completion of treatment and the child’s resilience and coping strategies have been linked to parental factors such as support as well as the parents’ own coping ability.94
Post-traumatic stress disorder (PTSD) is an important issue in all children heavily engaged in the healthcare system and relates to iatrogenic factors such as handling by multiple different clinicians, experiencing pain, separation from parents and undergoing procedures (e.g. phlebotomy, imaging) against the infant’s will, with severe developmental and psychosocial implications later in life. Indeed, 10% of children admitted to an intensive care unit were found to develop PTSD, with parental stress reactions as the strongest correlated predictor,95 highlighting again the importance of addressing psychosocial issues within the whole family. Unsurprisingly, psychosocial outcomes relating to self-image and resilience are also influenced by parental response and resilience.94
Evidence on behavioural problems has been mixed: using the Child Behaviour Checklist, Becker at al., 2005 reported significant differences between children with craniosynostosis and the general population;96 whereas Van der Vlugt et al., 2009 found no difference to the general population when accounting for IQ.97 At school age, Kelleher et al., 2006 found that in children with nonsyndromic trigonocephaly, 33% required assessment by a school psychologist; 47% required remedial or resource hours; 20% required a special needs classroom due to behaviour issues; and 37% were reported to have behavioural issues such as attention deficit disorder, autism and hyperactivity by their parents.98
In later school life and adolescence, issues pertain to stigma and bullying, with a third of craniosynostosis patients experiencing this.64 Most cope sufficiently but continued support is important, with social skills interventions proving beneficial.99 Another issue arising in adolescent patients is autonomy to make decisions relating to treatment as they reach the age required for consent: it is of vital importance for them to be involved in the decision-making process in order to optimise their cooperation and satisfaction.100 Furthermore, it is critical that adolescents have realistic expectations of treatment.
Although there have been no studies following up nonsyndromic craniosynostosis patients for psychosocial problems in adulthood, some have identified psychosocial problems in adults with syndromic craniosynostosis. Relative to controls, adults with Apert and Crouzon syndromes had a lower level of education, were less often married, experienced less sexual relationships and more commonly had periods of depressive mood, but were as likely to report a positive attitude to life as controls.101,102 Some adults with non-surgically treated craniosynostosis reported such pronounced psychological problems that they were willing to undergo correction in adulthood, a fundamentally more complicated operation than in infants.103
Treatment
The surgical treatment of patients with syndromic craniosynostosis was developed in Paris in the early 1970s by Tessier104 and then later by Marchac and Renier.105 Surgery had a 2-fold aim: to achieve an enlargement of the cranial volume so as to prevent sequelae of ICP (e.g. developmental delay, visual impairment, etc.), and the correction of morphologic abnormalities of the cranium, the orbits, and the upper jaw.
Since the first surgical intervention for craniosynostosis, a great many surgical techniques for the various types of craniosynostosis have been described and it must be emphasized that there is no consensus on the optimal surgical techniques for skull reconstruction in any form of craniosynostosis.26
However, a broad distinction can be made between “passive” techniques and “active” remodelling procedures (see Figure 4). Passive methods involve resection of bone, thereby allowing the developing and expanding brain to modify the skull shape (with or without assistance of a moulding helmet). As can be seen from a standard head circumference chart the first few months of life are associated with the greatest rate of skull growth (due to rapid brain growth) – most skull growth occurring in the first 2 years of life. More recently, these passive techniques have been further refined by minimally invasive techniques which are associated with smaller skin incisions and the need for less blood transfusions.9 Such techniques include endoscopic strip craniectomy (+/- moulding helmet) as pioneered by Jimenez106,107 or the use of spring distraction.108-115
The active remodelling techniques, on the other hand, do not rely on the self-correcting capability, but attempt to obtain the desired skull shape by direct reconstruction (often utilising rigid fixation using absorbable plates and screw).64 This type of surgery can also be broadly divided into that used to correct sagittal synostosis116 (Figure 5) and that used to treat metopic (Figure 6) and coronal synostosis (Figure 7) – which usually involves a fronto-orbital advancement and remodelling (FOAR). This latter procedure requires the orbital bar to also be removed as well as the abnormal area of the front of the skull.

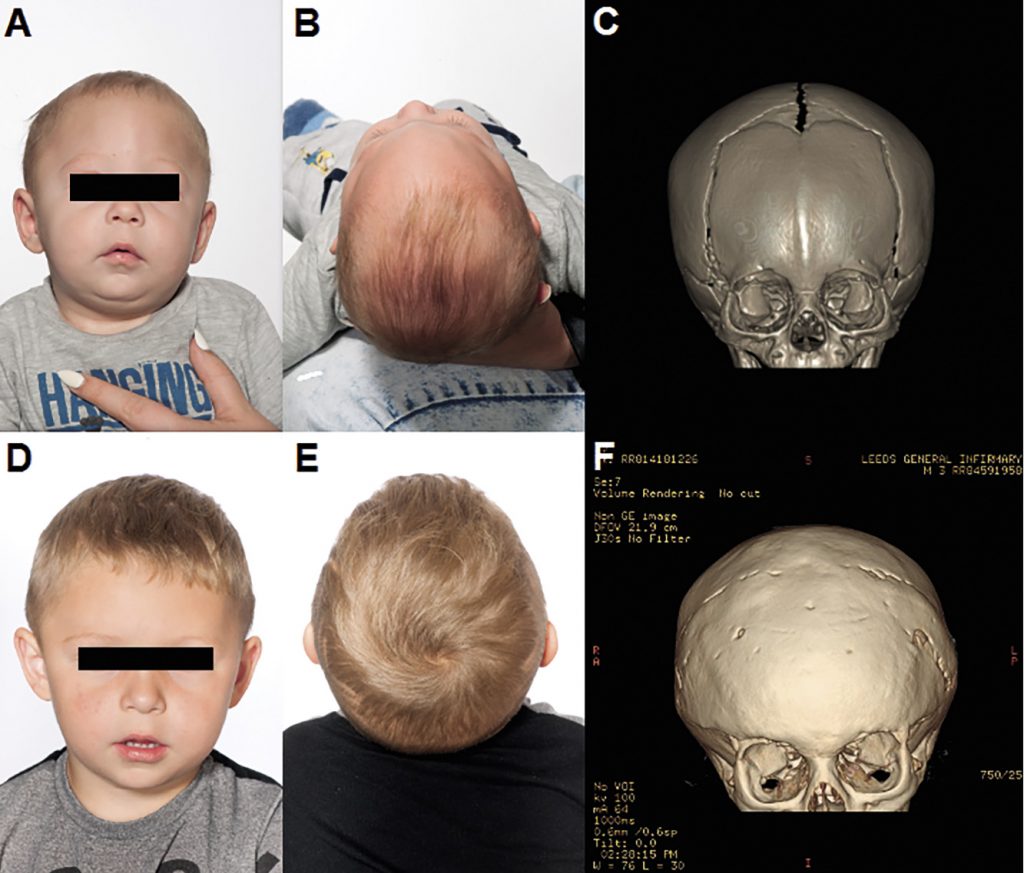
F: post-operative CT 3D Reconstruction.
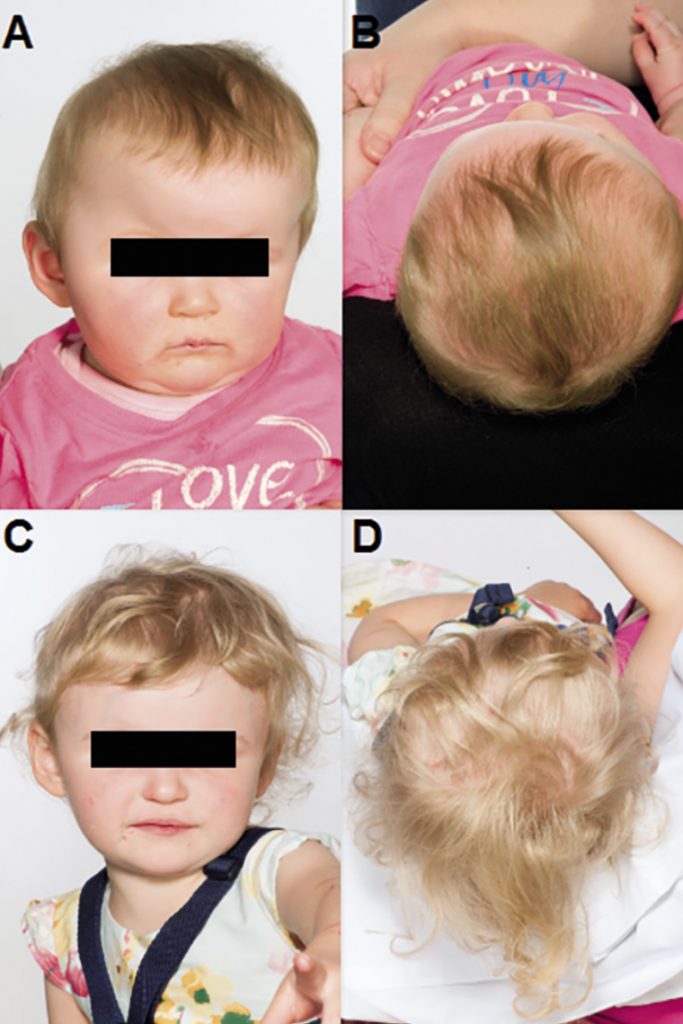
It has to be considered that in contrast to open craniosynostosis correction surgeries, which are generally performed between the ages of 6 to 18 months, minimally invasive procedures are performed much earlier within the first 3-6 months of age requiring early diagnosis and referral.117,118
However, the best surgical treatment has to be evaluated by the surgeon for each individual. Furthermore, especially in more complex syndromic craniosynostosis more than one surgery may be required.
a. Early intervention: Endoscopic or open strip craniectomy or spring distraction +/- post-operative orthotic helmet
vs.
b. Later intervention: Open calvarial reconstruction
Conclusions
The identification of the underlying genetic mutations and molecular mechanisms in craniosynostoses has led to a breakthrough in our understanding of these pathologies. A variety of procedures may be used to correct the deformity but over recent decades there has been increasing interest in early minimally invasive interventions where possible. Therefore, early diagnosis of craniosynostosis is imperative.
A multidisciplinary team approach in children with craniosynostosis and offering support to the entire family, including the parents, remains a vital factor in management of children with these pathologies. Long-term follow-up is particularly important as these children may encounter various problems throughout different stages in their development, including school age, adolescence and even further into early adulthood. Also, arising cognitive difficulties in non-syndromic craniosynostoses may be very subtle.119 Consequently, children will benefit from continuous assessments throughout childhood and early adulthood and in this way neuropsychological issues can be discussed and addressed accordingly.
References
- Lajeunie E, Le Merrer M, Bonaïti-Pellie C, Marchac D, Renier D. Genetic study of scaphocephaly. Am J Med Genet. 1996;62(3):282-5.
- Slater BJ, Lenton KA, Kwan MD, Gupta DM, Wan DC, Longaker MT. Cranial sutures: a brief review. Plast Reconstr Surg. 2008;121(4):170e-8e.
- Di Rocco F, Arnaud E, Renier D. Evolution in the frequency of nonsyndromic craniosynostosis. J Neurosurg Pediatr. 2009;4(1):21-5.
- Burokas L. Craniosynostosis: caring for infants and their families. Crit Care Nurse. 2013;33(4):39-50; quiz 1.
- Pyo, D., and Persing, J. A. Craniosynostosis. In SJ Aston, RW Beasley, and CHM Thorne (Eds.), Grabb and Smith’s Plastic Surgery. Philadelphia: Lippincott-Raven; 1997. P. 284.
- Renier D, Sainte-Rose C, Marchac D, Hirsch JF. Intracranial pressure in craniostenosis. J Neurosurg. 1982;57(3):370-7.
- Derderian C, Seaward J. Syndromic craniosynostosis. Semin Plast Surg. 2012;26(2):64-75.
- Clayman MA, Murad GJ, Steele MH, Seagle MB, Pincus DW. History of craniosynostosis surgery and the evolution of minimally invasive endoscopic techniques: the University of Florida experience. Ann Plast Surg. 2007;58(3):285-7.
- Proctor MR. Endoscopic craniosynostosis repair. Transl Pediatr. 2014;3(3):247-58.
- Anantheswar YN, Venkataramana NK. Pediatric craniofacial surgery for craniosynostosis: Our experience and current concepts: Part -1. J Pediatr Neurosci. 2009;4(2):86-99.
- McCarthy JG, Glasberg SB, Cutting CB, Epstein FJ, Grayson BH, Ruff G, et al. Twenty-year experience with early surgery for craniosynostosis: II. The craniofacial synostosis syndromes and pansynostosis–results and unsolved problems. Plast Reconstr Surg. 1995;96(2):284-95; discussion 96-8.
- Alvarez-Garijo JA, Cavadas PC, Vila MM, Alvarez-Llanas A. Sagittal synostosis: results of surgical treatment in 210 patients. Childs Nerv Syst. 2001;17(1-2):64-8.
- Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241(1):106-16.
- Badve CA, K MM, Iyer RS, Ishak GE, Khanna PC. Craniosynostosis: imaging review and primer on computed tomography. Pediatr Radiol. 2013;43(6):728-42; quiz 5-7.
- Weinzweig J, Kirschner RE, Farley A, Reiss P, Hunter J, Whitaker LA, Bartlett SP. Metopic synostosis: Defining the temporal sequence of normal suture fusion and differentiating it from synostosis on the basis of computed tomography images. Plast Reconstr Surg. 2003;112(5):1211-8.
- Vu HL, Panchal J, Parker EE, Levine NS, Francel P. The timing of physiologic closure of the metopic suture: a review of 159 patients using reconstructed 3D CT scans of the craniofacial region. J Craniofac Surg. 2001;12(6):527-32.
- Cohen MM, MacLean RE. Craniosynostosis: Diagnosis, Evaluation, and Management. 2 ed. Baltimore, MD: JHU Press; 2000.
- Persing JA, Jane JA, Shaffrey M. Virchow and the pathogenesis of craniosynostosis: a translation of his original work. Plast Reconstr Surg. 1989;83(4):738-42.
- Senarath-Yapa K, Chung MT, McArdle A, Wong VW, Quarto N, Longaker MT, et al. Craniosynostosis: molecular pathways and future pharmacologic therapy. Organogenesis. 2012;8(4):103-13.
- Rice DP. Craniofacial sutures. Development, disease and treatment. Preface. Front Oral Biol. 2008;12:xi.
- Selber J, Reid RR, Chike-Obi CJ, Sutton LN, Zackai EH, McDonald-McGinn D, et al. The changing epidemiologic spectrum of single-suture synostoses. Plast Reconstr Surg. 2008;122(2):527-33.
- van der Meulen J, van der Hulst R, van Adrichem L, Arnaud E, Chin-Shong D, Duncan C, et al. The increase of metopic synostosis: a pan-European observation. J Craniofac Surg. 2009;20(2):283-6.
- Kolar J. An Epidemiological Study of Nonsyndromal Craniosynostoses. J Craniofac Surg. 2011;22(1):47-49.
- Anderson IA, Goomany A, Bonthron DT, Bellew M, Liddington MI, Smith IM, et al. Does patient ethnicity affect site of craniosynostosis? J Neurosurg Pediatr. 2014;14(6):682-7.
- Kweldam CF, van der Vlugt JJ, van der Meulen JJ. The incidence of craniosynostosis in the Netherlands, 1997-2007. J Plast Reconstr Aesthet Surg. 2011;64(5):583-8.
- Garza RM, Khosla RK. Nonsyndromic craniosynostosis. Semin Plast Surg. 2012;26(2):53-63.
- Moore MH, Abbott AH, Netherway DJ, Menard R, Hanieh A. Bilambdoid and posterior sagittal synostosis: The Mercedes Benz syndrome. J Craniofac Surg. 1998;9:417–22.
- Feijen M, Franssen B, Vincken N, van der Hulst RR. Prevalence and Consequences of Positional Plagiocephaly and Brachycephaly. J Craniofac Surg. 2015;26(8):e770-3.
- Persing J, James H, Swanson J, Kattwinkel J, American Academy of Pediatrics Committee on Practice and Ambulatory Medicine ScoPSaSoNS. Prevention and management of positional skull deformities in infants. American Academy of Pediatrics Committee on Practice and Ambulatory Medicine, Section on Plastic Surgery and Section on Neurological Surgery. Pediatrics. 2003;112(1 Pt 1):199-202.
- Lennartsson F, Nordin P, Wennergren G. Teaching Parents How to Prevent Acquired Cranial Asymmetry in Infants. J Pediatr Nurs. 2016;31(4):e252-61.
- Guide I. Evaluation of Positional Plagiocephaly in Childrenguide: ISPN; [Available from: https://www.ispn.guide/.
- Governale LS. Craniosynostosis. Pediatr Neurol. 2015;53(5):394-401.
- Wilbrand JF, Lautenbacher N, Pons-Kühnemann J, Streckbein P, Kähling C, Reinges MH, et al. Treated Versus Untreated Positional Head Deformity. J Craniofac Surg. 2016;27(1):13-8.
- Ho JP, Mallitt KA, Jacobson E, Reddy R. Use of external orthotic helmet therapy in positional plagiocephaly. J Clin Neurosci. 2016;29:46-51.
- van Wijk RM, van Vlimmeren LA, Groothuis-Oudshoorn CG, Van der Ploeg CP, Ijzerman MJ, Boere-Boonekamp MM. Helmet therapy in infants with positional skull deformation: randomised controlled trial. BMJ. 2014;348:g2741.
- Chumas PD, Cinalli G, Arnaud E, Marchac D, Renier D. Classification of previously unclassified cases of craniosynostosis. J Neurosurg. 1997;86(2):177-81.
- Blount JP, Louis RG, Tubbs RS, Grant JH. Pansynostosis: a review. Childs Nerv Syst. 2007;23(10):1103-9.
- Lajeunie E, Le Merrer M, Bonaïti-Pellie C, Marchac D, Renier D. Genetic study of scaphocephaly. Am J Med Genet. 1996;62(3):282-5.
- Mulliken JB, Gripp KW, Stolle CA, Steinberger D, Müller U. Molecular analysis of patients with synostotic frontal plagiocephaly (unilateral coronal synostosis). Plast Reconstr Surg. 2004;113(7):1899-909.
- Wilkie AO, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJ, et al. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics. 2010;126(2):e391-400.
- Cohen MM. Etiopathogenesis of craniosynostosis. Neurosurg Clin N Am. 1991;2(3):507-13.
- Lajeunie E, Barcik U, Thorne JA, El Ghouzzi V, Bourgeois M, Renier D. Craniosynostosis and fetal exposure to sodium valproate. J Neurosurg. 2001;95(5):778-82.
- Richman JM, Lee SH. About face: signals and genes controlling jaw patterning and identity in vertebrates. Bioessays. 2003;25(6):554-68.
- Thomson A, Lotze M. The Cytokine Handbook, Two-Volume Set. 4 ed. London/ San Diego: Gulf professional Publishing; 2003.
- Schüller AC, Ahmed Z, Ladbury JE. Extracellular point mutations in FGFR2 result in elevated ERK1/2 activation and perturbation of neuronal differentiation. Biochem J. 2008;410(1):205-11.
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev. 1994;8(24):3032-44.
- Xu X, Weinstein M, Li C, Naski M, Cohen RI, Ornitz DM, et al. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development. 1998;125(4):753-65.
- Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235-53.
- De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127(3):483-92.
- Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N, et al. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002;16(7):870-9.
- Maeno T, Moriishi T, Yoshida CA, Komori H, Kanatani N, Izumi S, et al. Early onset of Runx2 expression caused craniosynostosis, ectopic bone formation, and limb defects. Bone. 2011;49(4):673-82.
- Komori T. Signaling networks in RUNX2-dependent bone development. J Cell Biochem. 2011;112(3):750-5.
- Lee MS, Lowe GN, Strong DD, Wergedal JE, Glackin CA. TWIST, a basic helix-loop-helix transcription factor, can regulate the human osteogenic lineage. J Cell Biochem. 1999;75(4):566-77.
- Gripp KW, Zackai EH, Stolle CA. Mutations in the human TWIST gene. Hum Mutat. 2000;15(5):479.
- Paznekas WA, Cunningham ML, Howard TD, Korf BR, Lipson MH, Grix AW, et al. Genetic heterogeneity of Saethre-Chotzen syndrome, due to TWIST and FGFR mutations. Am J Hum Genet. 1998;62(6):1370-80.
- Zhao H, Feng J, Ho TV, Grimes W, Urata M, Chai Y. The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nat Cell Biol. 2015;17(4):386-96.
- Crouzon MO. La dysostose cranio-faciale héréditaire. In Bulletins Et Mémoires De La
Société D’anthropologie De Paris; 1935. - Gault DT, Renier D, Marchac D, Jones BM. Intracranial pressure and intracranial volume in children with craniosynostosis. Plast Reconstr Surg. 1992;90(3):377-81.
- Cinalli G, Renier D, Sebag G, Sainte-Rose C, Arnaud E, Pierre-Kahn A. Chronic tonsillar herniation in Crouzon’s and Apert’s syndromes: the role of premature synostosis of the lambdoid suture. J Neurosurg. 1995;83(4):575-82.
- Cohen MM, Kreiborg S. Growth pattern in the Apert syndrome. Am J Med Genet. 1993;47(5):617-23.
- Muenke M, Schell U, Hehr A, Robin NH, Losken HW, Schinzel A, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet. 1994;8(3):269-74.
- Cohen MM. Pfeiffer syndrome update, clinical subtypes, and guidelines for differential diagnosis. Am J Med Genet. 1993;45(3):300-7.
- Foo R, Guo Y, McDonald-McGinn DM, Zackai EH, Whitaker LA, Bartlett SP. The natural history of patients treated for TWIST1-confirmed Saethre-Chotzen syndrome. Plast Reconstr Surg. 2009;124(6):2085-95.
- Mathijssen IM. Guideline for Care of Patients With the Diagnoses of Craniosynostosis: Working Group on Craniosynostosis. J Craniofac Surg. 2015;26(6):1735-807.
- Edmond JC, Nischal K, Forbes B, Katowitz W, Costakos D. Update on the management of patients with craniostenosis. Journal of American Association for Pediatric Ophthalmology and Strabismus {JAAPOS}. 2011;15(1):e35.
- Hill CA, Vaddi S, Moffitt A, Kane AA, Marsh JL, Panchal J, et al. Intracranial volume and whole brain volume in infants with unicoronal craniosynostosis. Cleft Palate Craniofac J. 2011;48(4):394-8.
- Sgouros S, Hockley AD, Goldin JH, Wake MJ, Natarajan K. Intracranial volume change in craniosynostosis. J Neurosurg. 1999;91(4):617-25.
- Cinalli G, Sainte-Rose C, Kollar EM, Zerah M, Brunelle F, Chumas P, et al. Hydrocephalus and craniosynostosis. J Neurosurg. 1998;88(2):209-14.
- Thompson DN, Harkness W, Jones B, Gonsalez S, Andar U, Hayward R. Subdural intracranial pressure monitoring in craniosynostosis: its role in surgical management. Childs Nerv Syst. 1995;11(5):269-75.
- Aldridge K, Marsh JL, Govier D, Richtsmeier JT. Central nervous system phenotypes in craniosynostosis. J Anat. 2002;201(1):31-9.
- Aldridge K, Kane AA, Marsh JL, Yan P, Govier D, Richtsmeier JT. Relationship of brain and skull in pre- and postoperative sagittal synostosis. J Anat. 2005;206(4):373-85.
- Aldridge K, Kane AA, Marsh JL, Panchal J, Boyadjiev SA, Yan P, et al. Brain morphology in nonsyndromic unicoronal craniosynostosis. Anat Rec A Discov Mol Cell Evol Biol. 2005;285(2):690-8.
- Shillito J, Matson DD. Craniosynostosis: a review of 519 surgical patients. Pediatrics. 1968;41(4):829-53.
- Strahle J, Muraszko KM, Buchman SR, Kapurch J, Garton HJ, Maher CO. Chiari malformation associated with craniosynostosis. Neurosurg Focus. 2011;31(3):E2.
- Noetzel MJ, Marsh JL, Palkes H, Gado M. Hydrocephalus and mental retardation in craniosynostosis. J Pediatr. 1985;107(6):885-92.
- Collmann H, Sörensen N, Krauss J. Hydrocephalus in craniosynostosis: a review. Childs Nerv Syst. 2005;21(10):902-12.
- Hoffman HJ, Hendrick EB. Early neurosurgical repair in craniofacial dysmorphism. J Neurosurg. 1979;51(6):796-803.
- Sainte-Rose C, LaCombe J, Pierre-Kahn A, Renier D, Hirsch JF. Intracranial venous sinus hypertension: cause or consequence of hydrocephalus in infants? J Neurosurg. 1984;60(4):727-36.
- Birgfeld CB, Dufton L, Naumann H, Hopper RA, Gruss JS, Haberkern CM, et al. Safety of Open Cranial Vault Surgery for Single-Suture Craniosynostosis: A Case for the Multidisciplinary Team. J Craniofac Surg. 2015;26(7):2052-8.
- Komotar RJ, Zacharia BE, Ellis JA, Feldstein NA, Anderson RC. Pitfalls for the pediatrician: positional molding or craniosynostosis? Pediatr Ann. 2006;35(5):365-75.
- Feijen MM, Claessens EA, Dovens AJ, Vles JS, van der Hulst RR. [Babies with cranial deformity]. Ned Tijdschr Geneeskd. 2009;153:A368.
- Ridgway EB, Weiner HL. Skull deformities. Pediatr Clin North Am. 2004;51(2):359-87.
- Bredero-Boelhouwer H, Treharne LJ, Mathijssen IM. A triage system for referrals of pediatric skull deformities. J Craniofac Surg. 2009;20(1):242-5.
- Bredero-Boelhouwer H, Treharne LJ, Mathijssen IM. A triage system for referrals of pediatric skull deformities. J Craniofac Surg. 2009 Jan;20(1):242-5.
- Tenhagen M, Bruse JL, Rodriguez-Florez N, Angullia F, Borghi A, Koudstaal MJ, et al. Three-Dimensional Handheld Scanning to Quantify Head-Shape Changes in Spring-Assisted Surgery for Sagittal Craniosynostosis. J Craniofac Surg. 2016;27(8):2117-23.
- Benson ML, Oliverio PJ, Yue NC, Zinreich SJ. Primary craniosynostosis: imaging features. AJR Am J Roentgenol. 1996;166(3):697-703.
- Chadduck WM, Chadduck JB, Boop FA. The subarachnoid spaces in craniosynostosis. Neurosurgery. 1992;30(6):867-71.
- Bellew M, Liddington M, Chumas P, Russell J. Preoperative and postoperative developmental attainment in patients with sagittal synostosis: 5-year follow-up. J Neurosurg Pediatr. 2011;7(2):121-6.
- Knight SJ, Anderson VA, Spencer-Smith MM, Da Costa AC. Neurodevelopmental outcomes in infants and children with single-suture craniosynostosis: a systematic review. Dev Neuropsychol. 2014;39(3):159-86.
- Virtanen R, Korhonen T, Fagerholm J, Viljanto J. Neurocognitive sequelae of scaphocephaly. Pediatrics. 1999;103(4 Pt 1):791-5.
- Arnaud E, Renier D, Marchac D. Prognosis for mental function in scaphocephaly. J Neurosurg. 1995;83(3):476-9.
- Kapp-Simon KA. Mental development and learning disorders in children with single suture craniosynostosis. Cleft Palate Craniofac J. 1998;35(3):197-203.
- Kapp-Simon KA, Figueroa A, Jocher CA, Schafer M. Longitudinal assessment of mental development in infants with nonsyndromic craniosynostosis with and without cranial release and reconstruction. Plast Reconstr Surg. 1993;92(5):831-9; discussion 40-1.
- Broder HL. Using psychological assessment and therapeutic strategies to enhance well-being. Cleft Palate Craniofac J. 2001;38(3):248-54.
- Bronner MB, Knoester H, Bos AP, Last BF, Grootenhuis MA. Posttraumatic stress disorder (PTSD) in children after paediatric intensive care treatment compared to children who survived a major fire disaster. Child Adolesc Psychiatry Ment Health. 2008;2(1):9.
- Becker DB, Petersen JD, Kane AA, Cradock MM, Pilgram TK, Marsh JL. Speech, cognitive, and behavioral outcomes in nonsyndromic craniosynostosis. Plast Reconstr Surg. 2005;116(2):400-7.
- van der Vlugt JJ, van der Meulen JJ, Creemers HE, Willemse SP, Lequin ML, Okkerse JM. The risk of psychopathology in children with craniosynostosis. Plast Reconstr Surg. 2009;124(6):2054-60.
- Kelleher MO, Murray DJ, McGillivary A, Kamel MH, Allcutt D, Earley MJ. Behavioral, developmental, and educational problems in children with nonsyndromic trigonocephaly. J Neurosurg. 2006;105(5 Suppl):382-4.
- Kapp-Simon KA, McGuire DE, Long BC, Simon DJ. Addressing quality of life issues in adolescents: social skills interventions. Cleft Palate Craniofac J. 2005;42(1):45-50.
- Lefebvre A, Barclay S. Psychosocial impact of craniofacial deformities before and after reconstructive surgery. Can J Psychiatry. 1982;27(7):579-84.
- Tovetjärn R, Tarnow P, Maltese G, Fischer S, Sahlin PE, Kölby L. Children with Apert syndrome as adults: a follow-up study of 28 Scandinavian patients. Plast Reconstr Surg. 2012;130(4):572e-6e.
- Fischer S, Tovetjärn R, Maltese G, Sahlin PE, Tarnow P, Kölby L. Psychosocial conditions in adults with Crouzon syndrome: a follow-up study of 31 Swedish patients. J Plast Surg Hand Surg. 2014;48(4):244-7.
- Marchac D, Renier D, Arnaud E. Unoperated craniosynostosis patients: correction in adulthood. Plast Reconstr Surg. 2008;122(6):1827-38.
- Kaufman BA, Muszynski CA, Matthews A, Etter N. The circle of sagittal synostosis surgery. Semin Pediatr Neurol. 2004;11(4):243-8.
- Mehta VA, Bettegowda C, Jallo GI, Ahn ES. The evolution of surgical management for craniosynostosis. Neurosurg Focus. 2010;29(6):E5.
- Barone CM, Jimenez DF. Endoscopic craniectomy for early correction of craniosynostosis. Plast Reconstr Surg. 1999;104(7):1965-73; discussion 74-5.
- Jimenez DF, Barone CM, Cartwright CC, Baker L. Early management of craniosynostosis using endoscopic-assisted strip craniectomies and cranial orthotic molding therapy. Pediatrics. 2002;110(1 Pt 1):97-104.
- Lauritzen C, Sugawara Y, Kocabalkan O, Olsson R. Spring mediated dynamic craniofacial reshaping. Case report. Scand J Plast Reconstr Surg Hand Surg. 1998;32(3):331-8.
- Lauritzen CG, Davis C, Ivarsson A, Sanger C, Hewitt TD. The evolving role of springs in craniofacial surgery: the first 100 clinical cases. Plast Reconstr Surg. 2008;121(2):545-54.
- Borghi A, Schievano S, Rodriguez Florez N, McNicholas R, Rodgers W, Ponniah A, James G, Hayward R, Dunaway D, Jeelani NUO. Assessment of spring cranioplasty biomechanics in sagittal craniosynostosis patients. J Neurosurg Pediatr. 2017;20(5):400-409.
- David LR, Plikaitis CM, Couture D, Glazier SS, Argenta LC. Outcome analysis of our first 75 spring-assisted surgeries for scaphocephaly. J Craniofac Surg. 2010;21(1):3-9.
- Davis C, Windh P, Lauritzen CG. Spring expansion is influenced by cranial biomechanics. J Craniofac Surg. 2010;21(3):843-6.
- Davis C, Lauritzen CG. The biomechanical characteristics of cranial sutures are altered by spring cranioplasty forces. Plast Reconstr Surg. 2010;125(4):1111-8.
- de Faria Valle Dornelles R, Cardim VL, de Campos Fonseca Pinto AC, Alonso N. Skull base cephalometric changes in cranial expansion by springs. J Craniofac Surg. 2011;22(4):1496-501.
- Tunçbilek G, Kaykçoğlu A, Bozkurt G, Akalan N. Spring-mediated cranioplasty in patients with multiple-suture synostosis and cloverleaf skull deformity. J Craniofac Surg. 2012;23(2):374-7.
- Mackenzie KA, Davis C, Yang A, MacFarlane MR. Evolution of surgery for sagittal synostosis: the role of new technologies. J Craniofac Surg. 2009;20(1):129-33.
- Proctor MR. Endoscopic cranial suture release for the treatment of craniosynostosis–is it the future? J Craniofac Surg. 2012;23(1):225-8.
- Stelnicki EJ. Endoscopic treatment of craniosynostosis. Atlas Oral Maxillofac Surg Clin North Am. 2002;10(1):57-72.
- Bellew M, Chumas P. Long-term developmental follow-up in children with nonsyndromic craniosynostosis. J Neurosurg Pediatr. 2015;16(4):445-51.