
Summary
- There is a major unmet need for therapies that slow or stop neuronal cell death in neurodegenerative disorders including PD. Conventional methods of developing new therapies are expensive and have yielded little.
- Innovations in trial design such as the use of re-purposed therapies in studies with multiple arms show much promise
- Better modeling of PD progression through Natural History studies will contribute to improved trial design.
The search for therapies capable of modifying, slowing or stopping neuronal cell death in neurodegenerative disorders remains elusive. In Parkinson’s disease (PD) a range of agents have been studied as potential disease modifying therapies (DMTs), including oral agents, and neurotrophic factors and genetransfection therapies delivered directly to the striatum. Transplants of tissue derived from foetal ventral mesencephalon have also been used with variable outcomes1. It is envisaged that a better understanding of cellular reprogramming events will soon herald an exciting new era of cellbased therapies for PD, with diseasemodification becoming a realistic proposition.
{kind=link}
However, studying DMT effects in PD is far from straightforward. Historically, the promise shown by many agents in phase II trials has not translated into benefits in larger phase III studies. Fundamental flaws in the design of these trials has contributed to this failure rate. In this article I outline the issues which we face in conducting trials of DMTs with particular reference to PD, and explore how innovations in clinical trial conduct and design might aid our evaluation of this exciting new generation of therapies.
Selection of candidate therapies for DMT trials
The conventional approach to developing a novel therapy is summarised in Figure 1. There are three main stages: Discovery (target identification, identification of lead compounds through screening, optimisation of lead compound), preclinical evaluation (pharmacological efficacy, evaluation of toxicology and interactions) and clinical development (phase I, II and III trials). Whilst this approach appeals to the scientific rationalist in us, it is timeconsuming and costly and has yielded few if any successes in Neurology, and none in the area of DMT.
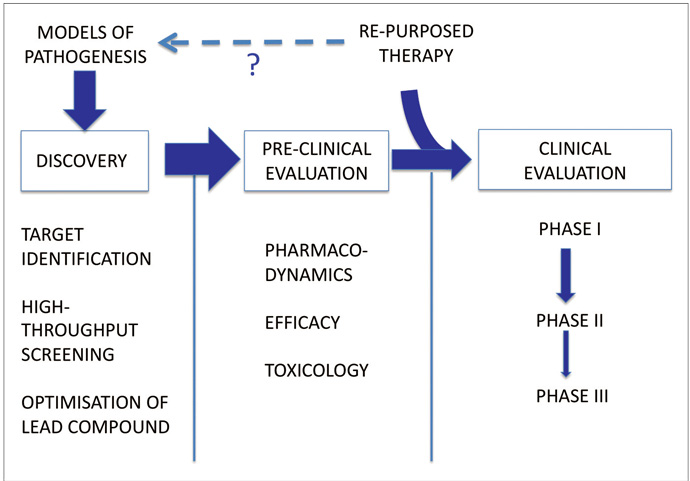
Alternative strategies must be considered. The repurposing or repositioning of drugs already approved in other indications is an increasingly popular approach.2 The principle is that biological active compounds approved in other indications may have additional ‘offtarget’ effects, including neuroprotective effects. By focusing upon agents with existing regulatory approval and safety data we can circumvent some of the cost and time constraints associated with drug development, and in some fields the success rates for repositioned drugs approaches 30%.3
Trials of repositioned drugs are already taking place in Neurology. Dimethyl Fumarate and, more recently, Simvastatin have shown effects in Multiple Sclerosis.4,5 In PD, a number of such studies are ongoing.6 Exenatide, originally developed as an antidiabetic drug, has been reported to show DMT properties in a small, openlabel ‘learning trial’.7 On the strength of such studies collaborative initiatives to advise on and coordinate learning trials of repurposed agents have been formed.6 Selection of appropriate candidates is the most critical and difficult aspect: criteria such as an ability to penetrate the bloodbrain barrier, effects in animal models and a proposed mode of action which accords with current understanding of PD pathogenesis could all reasonably be used. Scientifically this approach is less satisfying; the links with insights from basic science are weakened and, to an extent, it is hypothesisgenerating rather than hypothesistesting. Putative pathogenic mechanisms are invoked after the fact to explain observations, although it is plausible that useful insights into neurodegenerative mechanisms may be uncovered.
Trial design
Given that the natural history of treated PD typically runs for many years, trials of putative neuroprotective agents are likely to involve lengthy follow up periods in large sample groups. The costs of running such trials are considerable but could be mitigated, for example, by applying futility designs in pilot studies using smaller sample sizes, screening out therapies that are unlikely to prove effective. Futility designs typically involve the comparison of a single treatment arm with a predetermined lower limit of success (or an upper limit for worsening) in a one-sample test.8 Therapies performing the above criterion can then be selected for larger, phase III studies (“seamless” transition). Multiple futility studies can be run in parallel (multiarm or nested designs), including arms using combinations of therapies (Figure 2 below). Should one or more arms close, participants can be moved to alternative arms (adaptive design)? Efficient trial designs reduce turnaround time and trial costs.
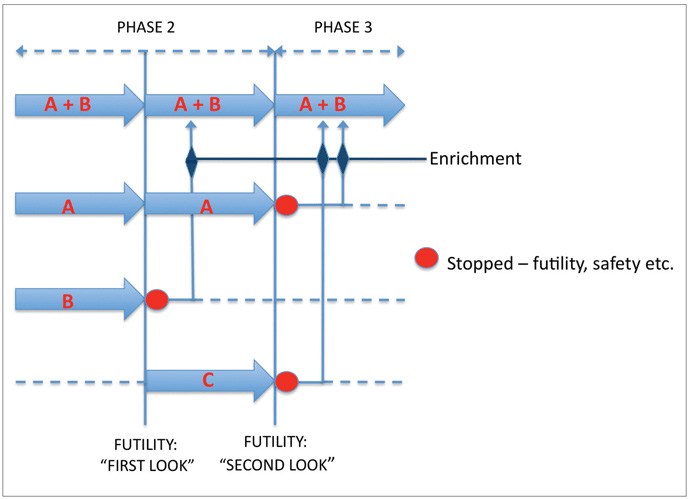
One problem inherent in the application of futility designs to the study of DMTs in neurodegeneration is that such a design assumes that the absence of short-term efficacy precludes longterm efficacy. Theoretically, an agent modifying neuronal cell death signals may show a slowly developing benefit or a ‘lag-benefit’ and may be screened out in a futility trial. We are faced with a Catch-22, as extending the follow-up periods in futility trials defeats their original purpose! Simple pragmatism, then, must prevail. Detection of a clinically meaningful effect in fewer than 6 months is largely implausible, and an agent showing no efficacy after 18 months is unlikely to thereafter, thus a time frame of 6-18 months would be reasonable for pilot DMT studies, with the caveat that agents suspected a priori to have delayed benefits are less suitable for such futility trials. Detection of relevant effects over such a timeframe would be facilitated by the inclusion of subjects at risk of more rapid disease progression, such as those older at disease onset. In the future, genotyping for genetic polymorphisms that influence disease progression, as has been described for the glucocerebrosidase9 should further assist with stratification of trial subjects. DMTs in neurodegenerative disorders should be most effective when employed early in the disease course and this must also inform the inclusion criteria used in these studies.
Separation of symptomatic and DMT effects
Identifying the optimum outcome measures for trials of putative neuroprotective agents in PD is far from straightforward. Biomarkers of progression, such as those derived from imaging, have been used but are insensitive measures of neurodegeneration.10 Commonly used clinimetric rating scales, such as the Unified Parkinson’s disease Rating Scale part III (UPDRS-3) are capable of capturing long-term changes over time, but do not measure all aspects of neurodegeneration.11 Furthermore, any putative neuroprotective agent which augments dopaminergic function may produce concurrent symptomatic benefits which cannot easily be disentangled from true diseasemodification. We have previously proposed the use of alternative outcome measures, such as time to significant, irreversible disease milestones (loss of postural reflexes, dementia).12 This would, of course, necessitate extended periods of followup. In this regard, improving our understanding of the natural history of treated PD would be highly beneficial. Theoretically, the observed progression of trial participants on a given index could be tracked against their expected disease trajectory derived from an individualised natural history model. Such an approach would allow us to separate important DMT effects more easily, and might ultimately obviate the need for separate control arms.
The use of clinimetric rating scales introduces a further issue, namely the extent to which objective responses on such scales translate into day-to-day benefits for patients. As clinicians, instinctively we hone in on p-values, in so doing prioritising [statistically] significant differences ahead of clinically important differences. The Minimal Clinical Difference (MCD) is the minimum change on a scale that can be recognised by a rater/experienced by a patient. For the UPDRS-3, the MCD is approximately 2.5.13 For reference, in the ADAGIO Study using rasagline, the mean difference between early and delayed start arms at 72 weeks was <2 points on the UPDRS -Total.14
Related to this is the need to ensure DMT trials are powered appropriately. A priori sample size calculations require an estimate of the anticipated effect size, which may be unknowable. An alternative strategy would be to accept a consensus MCD and base power calculations upon this. It is also necessary to allow for the fact that ‘early looks’ at the data –as would be required in nested or futility designs – reduce statistical power.
The placebo and “lessebo” effects in PD trials
It has been suggested that the placebo effect is more powerful in PD than in other disorders.15 There may be an additional confounder that we need to consider in PD: the socalled ‘lessebo’ effect. This term describes a phenomena that emerged from a metaanalysis of RCTS of dopamine agonists conducted by Mestre et al.16 The magnitude of the benefit of active drug (UPDRS-3 improvement) was reduced in studies that employed a placebo arm. In other words, if participants knew there was a chance of being assigned to placebo, the benefit of the active drug was reduced. Furthermore, the size of this ‘lessebo’ effect was proportional to the prior odds of being assigned to placebo. The use of control information derived from natural history models rather than employing a conventional control arm would be one method to reduce the effect of this potential confounder.
Conclusion
This review, though by no means comprehendsive, has highlighted the important challenges facing clinical trials of DMTs in PD, and by extension other neurodegenerative disorders. Perhaps the neurological community needs to set aside certain scientific prejudices. The selection of agents based on their ability to engage particular targets is inefficient and, I would argue, we must be more pragmatic in our approach. High throughput screening of compounds for DMT effects, including repurposed therapies, must take advantage of modern trial design innovations. Improved biomarkers of true disease progression should be sought and utilised, and better models of the natural evolution of the disorder with time must inform our selection of meaningful and relevant outcome measures for DMT trials.
This is an exciting era in PD therapeutics: International initiatives such as the cell-transplantation programme TransEuro serve testament to this.17 Proving the benefits of novel DMTs will require a systematic approach to clinical trial design. This remains a considerable challenge, but one which we are increasingly able to meet.
References
- Evans JR, Mason SL, Barker RA. Current status of clinical trials of neural transplantation in Parkinson’s disease. Prog Brain Res. 2012;200:169.98.
- Novac N. Challenges and opportunities of drug repositioning. Trends Pharmacol Sci. 2013 May;34(5):267.72.
- O’Connor KA, Roth BL. Finding new tricks for old drugs: an efficient route for public.sector drug discovery. Nat Rev Drug Discov. 2005 Dec;4(12):1005.14.
- Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo.controlled phase 3 study of oral BG.12 or glatiramer in multiple sclerosis. N Engl J Med. 2012 Sep;20;367(12):1087.97.
- Chataway J, Schuerer N, Alsanousi A, Chan D, Macmanus D, Hunter K, et al. Effect of high.dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS.STAT): a randomised, placebo.controlled, phase 2 trial. Lancet. 2014 Mar (published on-line 19/3/2014);18.
- Brundin P, Barker RA, Conn PJ, Dawson TM, Kieburtz K, Lees AJ, et al. Linked clinical trials – the development of new clinical learning studies in Parkinson’s disease using screening of multiple prospective new treatments. J Parkinsons Dis. 2013 Jan;1;3(3):231.9.
- Aviles.Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, et al. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest. 2013 Jun;123(6):2730.6.
- Herson J. Predictive probability early termination plans for phase II clinical trials. Biometrics. 1979 Dec 2013;35(4):775.83.
- Winder.Rhodes SE, Evans JR, Ban M, Mason SL, Williams.Gray CH, Foltynie T, et al. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain. 2013 Feb;136(Pt 2):392.9.
- Marras C, Lang AE. Outcome measures for clinical trials in Parkinson’s disease: achievements and shortcomings. Expert Rev Neurother. 2004 Nov;4(6):985.93.
- Elm JJ, Goetz CG, Ravina B, Shannon K, Wooten GF, Tanner CM, et al. A responsive outcome for Parkinson’s disease neuroprotection futility studies. Ann Neurol. 2005 Feb;57(2):197.203.
- Evans JR, Barker RA. Defining meaningful outcome measures in trials of disease.modifying therapies in Parkinson’s disease. Expert Opin Pharmacother. 2011 Jun;12(8):1249.58.
- Shulman LM, Gruber.Baldini AL, Anderson KE, Fishman PS, Reich SG, Weiner WJ. The clinically important difference on the unified Parkinson’s disease rating scale. Arch Neurol. 2010 Jan;67(1):64.70.
- Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med. 2009 Sep 24;361(13):1268.78.
- Goetz CG, Leurgans S, Raman R. Placebo-associated improvements in motor function: comparison of subjective and objective sections of the UPDRS in early Parkinson’s disease. Mov Disord. 2002 Mar;17(2):283.8.
- Mestre TA, Shah P, Marras C, Tomlinson G, Lang AE. Another face of placebo: the lessebo effect in Parkinson disease: meta-analyses. Neurology. 2014 Apr 22;82(16):1402.9.
- Allan LE, Petit GH, Brundin P. Cell transplantation in Parkinson’s disease: problems and perspectives. Curr Opin Neurol. 2010 Aug;23(4):426.32.