

Introduction
Motor neuron disease, in its commonest clinical form amyotrophic lateral sclerosis (ALS), is characterised by the degeneration of upper motor neurons (UMNs) of the corticospinal tract (CST) and lower motor neurons (LMNs) of the brainstem nuclei and spinal cord anterior horns [1]. It is recognised to have clinical, pathological, and, in cases with an intronic hexanucleotide repeat expansion in C9ORF72, genetic overlap with frontotemporal dementia (FTD) [2]. Half of MND patients die within three years of symptom onset, typically via respiratory failure. There is no specific test for MND, and diagnosis is based on clinical features, with an average delay from symptom onset to diagnosis of one year.
To ask “what is the cause of MND?” is to underestimate the emerging pathogenic complexity. Only 5% of cases report a family history. Cytoplasmic inclusions of the ubiquitinated 43 kDa transactive- region DNA-binding protein, TDP-43, superficially appear to be the unifying hallmark for the 95% who have apparently sporadic MND. However, TDP-43 inclusions are notably absent in the 20% of familial cases associated with mutations of SOD1, despite being clinically indistinguishable from other MND patients. Thus, there appear to be multiple discrete ‘tributaries’ flowing towards a common ‘waterfall’, beyond which there is a typically rapid, relatively selective motor neuronal degeneration. In the vast majority of apparently sporadic cases, MND is likely to involve multiple genetic influences operating at a low level individually, combined with as yet poorly-defined environmental factors.
The brain in MND
Case reports of MND with brain involvement beyond the motor cortex can be found in the early 20th Century literature, now recognised as MND associated with FTD, largely of the behavioural variant. Later post mortem studies confirmed wide-spread cerebral involvement in MND even without frank dementia [3]. A spectrum of cognitive dysfunc- tion, predominantly of a dysexecutive nature, and detectable in at least a third of MND cases without frank FTD, emerged over the following decades, cemented by the finding of the common TDP-43 histological signature [4].
Aberrant axonal transport is one of several major themes in MND pathogenesis [5], in which an LMN ‘dying back’ is then presumed to account for the consistent involvement of the CST, noted even in apparently clinically LMN-only cases. However, the early occurrence of cognitive impairment in some cases of ALS, and a rare UMN-only variant of MND (primary lateral sclerosis) are often associated with severe atrophy of the motor cortex and strikingly absent LMN pathology, support a top-down ‘dying forward’ process. Combined clinical and post mortem analysis supports a main focus of pathology at the site of symptom onset, with contiguous spread through UMN and LMN populations [6]. A clear primacy of one over other remains uncertain, with the increasing sense that the disease must be understood as a whole ‘system’ degeneration that includes the brain at some level in all cases.
Excitotoxicity and cortical hyperexcitability in MND
Raised levels of CSF glutamate, and abnormalities of glutamate transport and reuptake in human post mortem tissue as well as animal models of MND, underpin an excitotoxic theme of pathogenesis, in which over-stimulation of motor neurons results in eventual calcium-mediated cell death [5]. An alternative or contributory mechanism might be a reduction in inhibitory neuronal influences. Cortical and spinal cord interneuronal local circuits are found intimately associated with both the UMN and LMN populations respectively, and a diverse range of evidence supports a role for their involvement in pathogenesis [7]. Paired transcranial magnetic stimulation can be used to explore cortical inhibitory circuits, and studies in MND demonstrated a reduction in the normal inhibitory response seen at short interstimulus intervals [8]. An adapted threshold-tracking method to assess this reduction in short-interval intracortical inhibition has demonstrated specificity for MND over mimic disorders [9]. Furthermore, this methodology provided pivotal evidence for an increase in cortical hyperexcitability prior to the development of symptoms in carriers of SOD1 mutations [10].
Neuroimaging evidence for an ‘interneuronopathy’ in MND
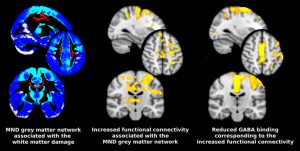
Pioneering in vivo cortical studies in MND employed activation positron emission tomography (PET), observing an extended area of cortical activation in response to a motor task. It was suggested this “boundary shift” might reflect loss of inhibitory local circuits [11]. Application of the PET ligand flumazenil confirmed widespread cerebral reductions in γ-aminobutyric acid (GABA)-A receptor binding in MND, crucially with relative preservation in those with a consistently more slowly progressive familial form of the disease (matched for disability and UMN involvement) [12].
Blood oxygenation level-dependent (BOLD) functional MRI involving a motor task, confirmed the original activation PET findings (reviewed in [13]). However, it is now possible to analyse the spontaneous fluctuations in BOLD signal that occur at rest (as opposed to during a motor task). This is being explored as a potentially sensitive in vivo marker of pathology in several neurodegenerative disorders. Diffusion tensor imaging (DTI) is an established MRI technique that can be used to define the characteristic structural white matter tract involvement in MND, which consistently involves the CST and corpus callosum [14], but also extra-motor frontal lobe pathways. The grey matter projections from these damaged white matter tracts were mapped using tractography in order to explore the resting-state functional changes in relation to an ‘MND- specific’ cortical network [15]. Unexpectedly this revealed increased functional connectivity within the structurally degenerating cortical network. Furthermore, this network overlapped strikingly with areas previously noted to show reduced GABA binding [12] (Figure). Whilst a ‘compensatory’ explanation cannot be entirely dismissed, it was noteworthy that those with faster rates of disease progression showed greater functional connectivity, raising the possibility that loss of inhibitory neuronal influences might directly contribute to the pathogenic cascade in MND.
Is there a potential MND brain architecture?
Osler noted that: “It is much more important to know what sort of a patient has a disease than what sort of disease a patient has.”Case-control studies of the strong anecdotal observation of pre-morbid athleticism among those developing MND have been inconclusive. The significantly increased occupational risk of MND associated with professional football and military service superficially fits with greater athleticism, and a significantly reduced vascular risk profile has also been noted among MND patients and relatives [16]. Debate continues over exercise as a direct precipitant of disease, versus athleticism as an associated phenotype. Developmental factors must also be considered, with MND patients noted to have a surrogate marker for high intrauterine testosterone exposure (also common among athletes) in the form of a reduced 2D:4D digit ratio [17].
With these data in mind, though entirely speculatively, it is conceivable that athleticism might in part reflect a particular cerebral architecture, which may, in turn, be a preferred substrate for MND in a minority. Eisen speculated on the relatively rapid human neocortical development associated with opposable thumbs and bipedalism as holding potential importance for the pathogenesis of MND [18]. Distinct patterns of structural and functional brain network organisation in healthy individuals have already been linked to the stereotyped patterns of neurodegenerative diseases [19]. Aptly summarised: “What wires together, dies together” [20].
Hand dominance has been linked to reduced inhibitory influences in the contralateral hemisphere, and so the finding of significantly increased concordance of the site of onset and handedness in those with upper limb-onset MND might reflect inhibitory, interneuronal organisation, rather than an activity-driven mechanism [21]. In this way, a less inhibited, or perhaps more functionally connected motor cortex, with all its evolutionary advantage in terms of physical performance in youth, might somehow be more vulnerable, perhaps more permissive of the spread of pathology, or otherwise define variable rates of progression in MND.
Primary prevention of MND?
Speculation aside, it is likely that the development of clinical symptoms in MND represents the late stages of long-standing pathological
processes. Indeed the primary reason for the failure of so many of the candidate therapies for MND may be their administration in a relatively intractable phase of pathology. Although a minority of cases, those asymptomatic individuals carrying single genetic mutations linked to the development of MND offer a valuable opportunity to study the earliest changes. The observation of a similar pattern of metabolic changes in the cervical spinal cord of presymptomatic SOD1 mutation carriers compared to those with established disease [22], marks a major conceptual shift in where pathology in MND begins, and brings it in line with observations in Alzheimer’s, Parkinson’s and Huntington’s Diseases.
High-field MR spectroscopy can now be used to specifically demonstrate reduced GABA within the motor cortex in MND patients [23]. In combination with other advanced MRI techniques, the refinement of a cortical ‘disinhibitory signature’ in presymptomatic cases will be an important step towards the long-term aspiration of primary prevention. For the larger apparently sporadic and symptomatic group, identifying such a signature has the potential to reduce diagnostic delay, and permit earlier administration of therapy, one strategy for which might be boosting inhibitory interneuronal function.
References
- Kiernan –MC, Vuc-ic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942-55.
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323-30.
- Smith MC. Nerve fibre degeneration in the brain in amyotrophic lateral sclerosis. JNeurolNeurosurgPsychiatry. 1960;23:269-82.
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al.
Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130-3. - Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65 Suppl 1:S3-9.
- Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009;73:805-11.
- Turner MR, Kiernan MC. Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotroph Lateral Scler. 2012;13:245-50.
- Yokota T, Yoshino A, Inaba A, Saito Y. Double cortical stimulation in amyotrophic lateral sclerosis. JNeurolNeurosurgPsychiatry. 1996;61:596-600.
- Vucic S, Cheah BC, Yiannikas C, Kiernan MC. Cortical excitability distinguishes ALS from mimic disorders. Clin Neurophysiol. 2011;122:1860-6.
- Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008;131:1540-50.
- Kew JJ, Leigh PN, Playford ED, Passingham RE, Goldstein LH, Frackowiak RS, et al Cortical function in amyotrophic lateral sclerosis. A positron emission tomography study. Brain. 1993;116 (Pt 3):655-80.
- Turner MR, Hammers A, Al-Chalabi A, Shaw CE, Andersen PM, Brooks DJ, et al. Distinct cerebral lesions in sporadic and ‘D90A’ SOD1 ALS: studies with [11C]flumazenil PET. Brain. 2005;128:1323-9.
- Turner MR, Agosta F, Bede P, Govind V, Lule D, Verstraete E. Neuroimaging in amyotrophic lateral sclerosis. Biomark Med. 2012;6:319-37.
- Filippini N, Douaud G, Mackay CE, Knight S, Talbot K, Turner MR. Corpus callosum involvement is a consistent feature of amyotrophic lateral sclerosis. Neurology. 2010;75:1645-52.
- Douaud G, Filippini N, Knight S, Talbot K, Turner MR. Integration of structural and functional magnetic resonance imaging in amyotrophic lateral sclerosis. Brain. 2011;134:3470-9.
- Turner MR, Wotton C, Talbot K, Goldacre MJ. Cardiovascular fitness as a risk factor for amyotrophic lateral sclerosis: indirect evidence from record linkage study. J Neurol Neurosurg Psychiatry. 2012;83:395-8.
- Vivekananda U, Manjalay ZR, Ganesalingam J, Simms J, Shaw CE, Leigh PN, et al. Low index-to-ring finger length ratio in sporadic ALS supports prenatally defined motor neuronal vulnerability. J Neurol Neurosurg Psychiatry. 2011;82:635-7.
- Eisen A. Amyotrophic lateral sclerosis-Evolutionary and other perspectives. Muscle Nerve. 2009;40:297-304.
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42-52.
- Bak TH, Chandran S. What wires together dies together: Verbs, actions and neurodegeneration in motor neuron disease. Cortex. 2012;48:936-44.
- Turner MR, Wicks P, Brownstein CA, Massagli MP, Toronjo M, Talbot K, et al. Concordance between site of onset and limb dominance in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2010;82:853-4.
- Carew JD, Nair G, Andersen PM, Wuu J, Gronka S, Hu X, et al. Presymptomatic spinal cord neurometabolic findings in SOD1-positive people at risk for familial ALS. Neurology. 2011;77:1370-5.
- Foerster BR, Callaghan BC, Petrou M, Edden RA, Chenevert TL, Feldman EL. Decreased motor cortex gamma-aminobutyric acid in amyotrophic lateral sclerosis. Neurology. 2012;78:1596-600.