

Introduction
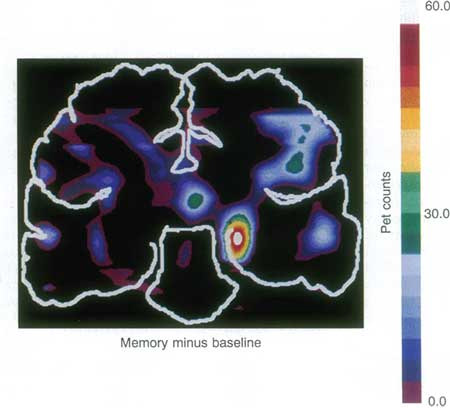
The hippocampus plays a fundamental role in learning and memory processes, as famously discovered with the study of patient HM, who lost the ability to lay down new memories after surgical removal of the medial temporal lobes [1]. Not only is the hippocampus necessary for memory formation, but it is also activated during memory tasks (Figure 1). The acquired information is thought to be stored as changes in the connections between hippocampal neurons and such changes are known as synaptic plasticity, the strengthening, and weakening of synaptic efficacy. The best-described hippocampal plasticity process is that of long-term potentiation (LTP), which will be discussed here.
Forty years ago, Bliss and Lømo reported that a high-frequency train of presynaptic stimulation in the rabbit hippocampus in vivo caused strengthening of the postsynaptic response to a given stimulation, [2] confirming earlier suggestions about activity-driven synaptic changes proposed independently by Konorski (1948) [3] and Hebb (1949) [4]. This “long-lasting potentiation” is now known as LTP and has received much experimental attention over the years due to the widely held view that it could be a cellular substrate for learning and memory. Initial exploratory research confirmed that LTP was found in other animals, other hippocampal pathways and brain regions, and even in vitro brain slice preparations, which proved particularly important for further detailed experimental dissection.
Thirty years ago, a fundamental step forward was made in our mechanistic understanding of LTP with the demonstration of NMDA receptor (NMDAR) involvement [5]. Upon activation by synaptically-released glutamate, NMDARs open an ion channel permeable to Ca2+, required for LTP [6]. Ten years later, Bliss and Collingridge summarised the intracellular pathways of plasticity that had been uncovered so far and outlined a number of crucial unresolved issues in their seminal review. [7] So how far have we come in answering them?
Major recent advances
The long-established mechanism of hippocampal LTP involves the presynaptic release of glutamate along with postsynaptic depolarisation sufficient to relieve the NMDAR of its Mg2+ block. The ensuing Ca2+ influx through NMDARs can then trigger downstream kinase cascades that cause the increased recruitment of AMPA receptors (AMPARs) to the postsynaptic membrane, which supports the initial increase in synaptic strength. In the longer term, changes in synaptic strength are maintained by synaptic remodelling. Significant progress has been made in our understanding of the mechanisms underlying the induction, expression, and maintenance of synaptic plasticity.
Induction
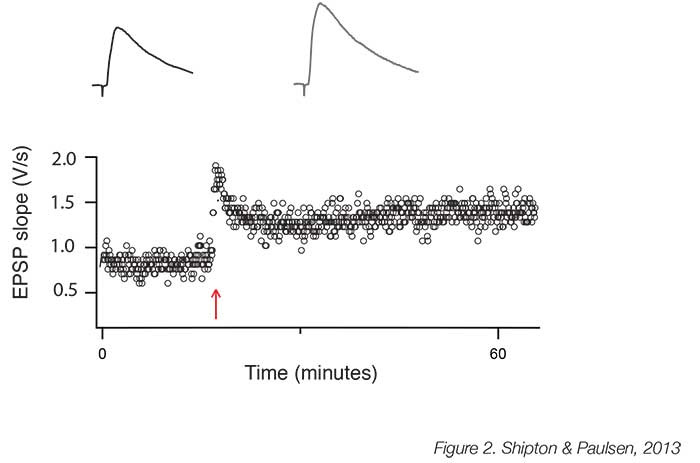
The necessity of NMDARs for the induction of hippocampal LTP is unrefuted; indeed, the crucial feature of LTP induction protocols is that they cause sufficient depolarisation to permit a high level of Ca2+ influx through these NMDARs. The induction protocols most commonly used are high-frequency (‘tetanic’) stimulation (Figure 2), theta burst stimulation and pre-postsynaptic spike pairing. The latter paradigm induces spike-timing-dependent potentiation (tLTP) [8], which confirms Hebb’s prediction that potentiation occurs when the presynaptic neuron takes part in firing the postsynaptic neuron [4]. tLTP is, therefore, an attractive model for learning as these precise timing requirements of neuronal activity indicate a mechanism for encoding associations in the external world. NMDARs comprise four subunits, two obligatory GluN1 subunits and two additional GluN2 subunits, of which there are different types. It is increasingly recognised that GluN2B subunit-containing NMDARs are required to support LTP [9], most likely because of the tight association between the GluN2B subunit and Ca2+/calmodulin-dependent protein kinase II, which has been identified as a crucial enzyme in LTP induction [10]. Interestingly, GluN2B subunit-containing NMDARs may be particularly vulnerable to, and help mediate, the synaptotoxic effects of amyloid beta [11], and so further understanding the unique role they play at the synapse and in LTP could help with the treatment of neurodegenerative diseases that affect learning and memory.
Expression
It is well established that a greater number of AMPARs at the postsynaptic membrane supports the increased synaptic strength in LTP. This process involves the initial recruitment of GluA1 subunit-containing AMPARs to the postsynaptic membrane and their subsequent replacement by GluA2- containing AMPARs to support a continued enhancement of synaptic strength. [12] However, the mechanism has recently emerged as more complex than the simple direct insertion of AMPARs at the post-synaptic site, and instead, it appears that a three-stage model may explain this increase [13]. Firstly, LTP triggers exocytosis of GluA1-containing AMPARs to extra-and peri-synaptic regions; secondly, these diffuse laterally into synaptic sites; thirdly, receptors must be captured by the scaffolding of the postsynaptic density in order to be maintained at the synapse. Although LTP pathways likely exert tight regulation of the initial membrane insertion stage, a recent paper suggests that the subsequent steps of this process may be more flexible. Granger et al found that, although a reserve pool of receptors was required to support synaptic strengthening in the hippocampus, the subtype identity of the AMPARs was unimportant; moreover, even the kainate receptor, a glutamate receptor not normally found at that synapse, could substitute.14 Although this research was conducted with endogenous AMPARs deleted, this newly found flexibility at the synapse has implications for the clinic since it opens the door to the introduction of new designer receptors to restore synaptic function in neurodegenerative conditions. However, this lack of specificity is a double-edged sword, since perturbing this process at a synapse could have many unforeseen consequences.
Maintenance
It has been suggested that LTP is maintained by persistent kinase activity. A strong candidate for this role appeared to be a kinase known as PKM. since blocking it with a peptide called ZIP reversed both hippocampal LTP and specific spatial memories [15]. However, the importance of this ‘memory molecule’ has recently been questioned as two labs have reported that hippocampal LTP and hippocampus-dependent learning are present in both constitutive and conditional PKM. knock-out mice [16,17]. In fact, it seems unlikely that a mechanism merely involving elevated kinase activity could maintain information storage over years, and a better candidate to sustain enhanced synaptic strength would be structural changes at the synapse. Imaging in neuronal cultures and in animals in vivo has revealed two major structural changes associated with LTP. Firstly, pre-existing thin postsynaptic spines enlarge. [18] Secondly, new spines appear, and LTP also correlates with the stabilisation of such nascent spines. [19] When spines enlarge sufficiently they become known as mushroom spines; chronic imaging has revealed that spines of this type can be stable for months [20] and are not permanently changed following an LTP protocol, [18] suggesting they may be robust to perturbations and hence able to act as a memory store’.The positive correlation between the volume of the posterior hippocampi of London taxi drivers and the length of time spent in this hippocampally-demanding career is an indication, although not without alternative interpretations, that long-term structural changes also exist in humans [21].
Plasticity and behaviour
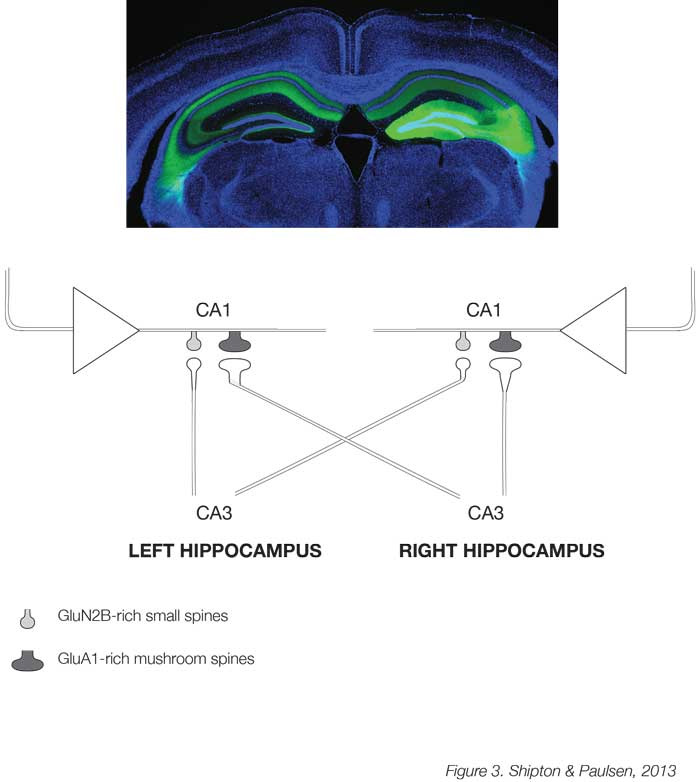
A major challenge for the future is to understand the relationship between plasticity and behaviour. Although the implicit and explicit assumption has been that plasticity supports memory [22], we still only have correlative evidence for this assertion, and, combined with the recent finding that NMDARs in the CA1 are not always necessary for hippocampus-dependent learning and memory [23], it remains unresolved. A newly developed technology, known as optogenetics, which allows acute control of neuronal activity in regionally-and genetically-defined cell populations by light, may help probe a causal link between plasticity and memory. This has already been used at the cellular level to show that reactivating neurons in the dentate gyrus of the hippocampus that were active during acquisition of fear conditioning triggers expression of the context-dependent fear memory [24]. The next stage is to investigate the relationship between plasticity and behaviour at the level of synapses. One promising model to investigate whether distributed changes in synaptic weights can support learning and memory may be the synapse between the excitatory neurons of the CA3 and CA1 regions in the mouse hippocampus. Here, an unexpected asymmetric distribution of postsynaptic spine types has been found whereby spines in CA1 neurons that receive input from the CA3 of the left hippocampus are mostly small and rich in GluN2B subunit-containing NMDARs, while those receiving right CA3 projections are larger and rich in AMPARs [25] (Figure 3). Due to this hemispheric asymmetry, these different types of spines can be targeted optogenetically and it was found that only the GluN2B-rich spines receiving left CA3 input show tLTP [26]. It is now possible to manipulate these two types of synapses with their different propensity for plasticity to test whether they have differential effects on learning and memory in behaving mice.
Clinical relevance
Synaptic dysfunction is a central component of a wide range of neurological conditions. Understanding the basic mechanisms involved in hippocampal LTP may make it possible to alter plasticity and neural networks in predictable ways, and the hope is that this could be used therapeutically in the future. For example, the early stages of Alzheimer’s disease are characterised by synapse loss in the hippocampus and the inability to lay down new memories. Using our greater understanding of the types of NMDARs and AMPARs involved in LTP induction and expression respectively, it might be envisaged that the synapse could be modified to prevent the initial synaptotoxicity and consequent neurodegeneration at early stages of the disease, or adapted to restore plasticity to remaining synapses later in the condition. One way to modify synapses to this end may be the introduction of NMDARs with a higher potential for LTP. Alternatively, so called ‘designed receptors exclusively activated by designer drugs’ (DREADDs) could be used. This novel pharmacogenetic technology involves stereotactic delivery of viral constructs containing DREADDs under specific promoter control so that engineered receptors are expressed by one type of neuron in a limited brain region, and then can be activated on demand by administration of a designer drug to stimulate their function, for example to enhance plasticity. It is also conceivable that downstream kinases could be targeted, but these may have more non-specific effects on cellular processes and so be less amenable to manipulation. Of course, the ability to restore learning without eradicating old memories is a huge challenge, but further study of the mechanisms that generate and maintain the aforementioned hippocampal asymmetry in the distribution of synapses with different propensities for plasticity could provide insight into how we may hope to achieve this in the future.
The possibility of genetically altering synaptic receptor composition in discrete brain regions has broader clinical ramifications than treating diseases of synaptic dysfunction. The ability to ‘reopen’ a brain region for plasticity may be particularly useful in aiding recovery from stroke or brain injury. Moreover, since both optogenetics and DREADDs enable the stimulation or suppression of activity in specific neuronal populations and circuits, they might prove a desirable replacement for deep brain stimulation in the treatment of Parkinson’s disease and depression. They could also be developed to treat obsessive-compulsive disorder, addiction and otherwise intractable epilepsies. For example, it was recently shown that both optogenetic and K+ channel gene therapy were successful treatments in a rat model of focal neocortical epilepsy [27]. Pharmacogenetics is a particularly appealing approach as, apart from the initial stereotactic surgery, it would be less invasive than chronic implantation of electrical or optical stimulation devices. Nevertheless, the ability to manipulate neuronal circuits at such a finely tuned level also brings with it many ethical considerations.
Conclusions
Huge advances have been made in the forty years since LTP was first formally described, though we are still unravelling the complexity of the various mechanisms of induction, expression and maintenance that support synaptic strengthening. The biggest scientific question in the field of whether or not LTP is the memory mechanism remains unanswered. Excitingly, novel technologies now make it possible to test a causal relationship to definitively answer this question, and, moreover, probe how exactly plasticity supports learning and memory. Furthermore, there is potential scope to harness our current knowledge for therapeutic purposes, not only in disorders affecting learning and memory but also to enhance recovery following brain injury.
References
- Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry. 1957;20:11-21.
- Bliss TV, Lømo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331-56.
- Konorski, J. Conditioned reflexes and neuron organization. New York: Cambridge University Press. 1948.
- Hebb, DO. The organization of behavior. New York: Wiley & Sons. 1949.
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33-46.
- Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719-21.
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31-9.
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464-72.
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081-94.
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289-301.
- Malinow R. New developments on the role of NMDA receptors in Alzheimer’s disease. Curr Opin Neurobiol. 2012;22:559-63.
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331-43.
- Opazo P, Choquet D. A three-step model for the synaptic recruitment of AMPA receptors. Mol Cell Neurosci. 2011;46:1-8.
- Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA. LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature. 2013;493:495-500.
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141-4.
- Lee AM, Kanter BR, Wang D, Lim JP, Zou ME, Qiu C, McMahon T, Dadgar J, Fischbach-Weiss SC, Messing RO. Prkcz null mice show normal learning and memory. Nature. 2013;493:416-9.
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. PKM-. is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420-3.
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761-6.
- Hill TC, Zito K. LTP-induced long-term stabilization of individual nascent dendritic spines. J Neurosci. 2013;33:678-86.
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279-91.
- Maguire EA, Gadian DG, Johnsrude IS, Good CD, Ashburner J, Frackowiak RS, Frith CD. Navigation-related structural change in the hippocampi of taxi drivers. Proc Natl Acad Sci U S A. 2000;97:4398-403.
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649-711.
- Bannerman DM, Bus T, Taylor A, Sanderson DJ, Schwarz I, Jensen V, Hvalby Ø, Rawlins JN, Seeburg PH, Sprengel R. Dissecting spatial knowledge from spatial choice by hippocampal NMDA receptor deletion. Nat Neurosci. 2012;15:1153-9.
- Liu X, Ramirez S, Pang PT, Puryear CB, Govindarajan A, Deisseroth K, Tonegawa S. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature. 2012;484:381-5.
- Shinohara Y, Hirase H, Watanabe M, Itakura M, Takahashi M, Shigemoto R. Left-right asym- metry of the hippocampal synapses with differential subunit allocation of glutamate receptors. Proc Natl Acad Sci U S A. 2008;105:19498-503.
- Kohl MM, Shipton OA, Deacon RM, Rawlins JN, Deisseroth K, Paulsen O. Hemisphere-specific optogenetic stimulation reveals left-right asymmetry of hippocampal plasticity. Nat Neurosci. 2011;14:1413-5.
- Wykes RC, Heeroma JH, Mantoan L, Zheng K, MacDonald DC, Deisseroth K, Hashemi KS, Walker MC, Schorge S, Kullmann DM. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med. 2012;4:161ra152.
- Squire LR, Ojemann JG, Miezin FM, Petersen SE, Videen TO, Raichle ME. Activation of the hippocampus in normal humans: A functional anatomical study of memory. Proc Natl Acad Sci U S A. 1992;89:1837-41.