

Summary
- Lysosomes are subcellular organelles responsible for the physiologic turnover of cell constituents. Lysosomes are involved not only in degradation, but also in fundamental processes such as secretion, plasma membrane repair, signalling and energy metabolism.
- Lysosomal storage diseases (LSDs) describe a heterogeneous group of rare inherited disorders characterised by the accumulation of undigested or partially digested macromolecules in the lysosomes. This accumulation disrupts the cell’s normal functioning and gives rise to the clinical manifestations of LSDs. LSDs affect different body organs or systems including the bones, eyes, heart, lungs, kidneys, skin, and frequently the central nervous system.
- Autophagy is a catabolic pathway through which cellular components (including dysfunctional organelles) are engulfed in vesicles (autophagosomes) and recycled upon fusion of autophagosomes with lysosomes. The block of autophagy (impaired fusion of autophagosomes with lysosomes) causes the accumulation of toxic material and contributes to neurodegeneration in LSDs. The impairment of autophagy initiates degenerative processes not only in LSDs but also in the most common form of age-related neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases.
- It has been demonstrated that α-synuclein, a presynaptic protein prone to misfolding, can aggregate and contribute to the pathogenesis of some neurodegenerative diseases through impaired autophagy capability in neuronal cells.
- Lysosomal-dependent α-synuclein accumulation found in LSDs might contribute to trigger neurodegeneration in these pathologies by causing α-synuclein chaperoning deficit and presynaptic failure.
Introduction
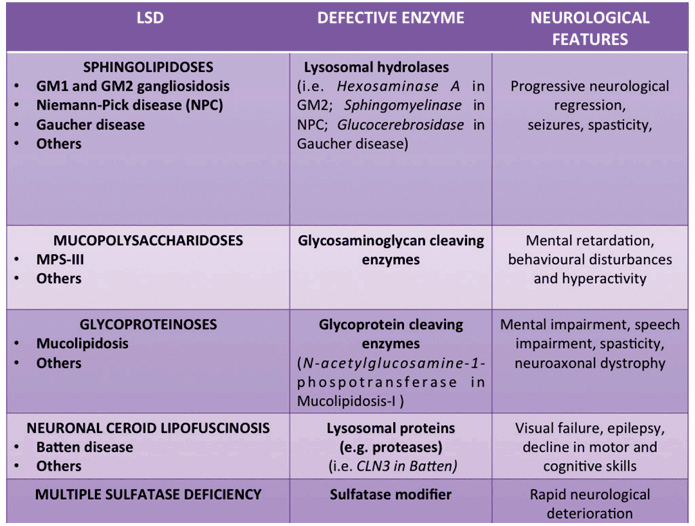
Lysosomal storage diseases (LSDs) are a family of disorders resulting from inherited gene mutations that perturb lysosomal homeostasis, thus ultimately leading to the accumulation of undegraded material into lysosomes.1 Although most LSDs result from acidic hydrolase deficiencies, a considerable number of these conditions result from defects in either membrane lysosomal proteins or in other non-lysosomal proteins that are critical for proper function of the lysosomal system. The incidence of LSDs is estimated to be approximately 1:5,000 live births, but the true figure is likely greater because of undiagnosed or misdiagnosed cases. The progressive lysosomal accumulation of undegraded metabolites results in lysosomal dysfunction and consequent generalised cell and tissue damage, and, ultimately, to multi-systemic pathology.2 Storage may begin during early embryonic development, and the clinical presentation for LSDs can vary from an early and severe phenotype to late-onset mild disease. Relatively few LSDs lack pathology in the central nervous system (CNS). Indeed, in the majority of LSDs, CNS involvement is common (Table 1) and the consequent symptoms are often the most debilitating because neurodegeneration can occur in multiple brain regions (e.g., thalamus, cortex, hippocampus, and cerebellum). In the context of improving the life expectancy of people with LSDs, new research initiatives have been launched with the specific remit of elucidating how lysosomal dysfunction may specifically affect neuronal function and viability, thus determining the neuropathological phenotype observed in LSDs. In this article we discuss the most recent advances in these studies useful for developing possible therapeutic interventions.
The Lysosome in normal cell physiology and diseases
Lysosomes are membrane-bound organelles with an acid lumen that contain several types of hydrolases that are responsible for the degradation of specific substrates. Lysosomal functions can be classified into three main types: degradation, secretion and signalling.3 Lysosomes are involved in the degradation and recycling of extracellular material (via endocytosis) and intracellular material (via autophagy). Extracellular material reaches the lysosome generally by endocytosis through specific endocytic mechanisms based on the nature of the cargo. Intracellular materials reach the lysosome through autophagy, a catabolic pathway used by cells to capture cytoplasmic components for degradation and recycling.4 Through autophagy, macromolecules and organelles are recycled via autophagosome-mediated transport to, and fusion with, lysosomes. The resulting breakdown products are used to generate new cellular components and energy in response to the nutritional needs of the cell. Lysosomes also undergo Ca2+ regulated exocytosis to secrete their content into the extracellular space and to repair damaged plasma membranes.5 Lysosomal exocytosis may also directly modulate cellular clearance.6 More recently, lysosomes have been identified as signalling organelles that can sense nutrient availability and activate a lysosome-to-nucleus signalling pathway that mediates the starvation response and regulates energy metabolism.7 The importance of proper lysosomal function to normal cell physiology is highlighted by the fact that lysosomal dysfunction initiates degenerative processes in a number of human diseases and is also involved in the process of ageing8 (Figure 1).
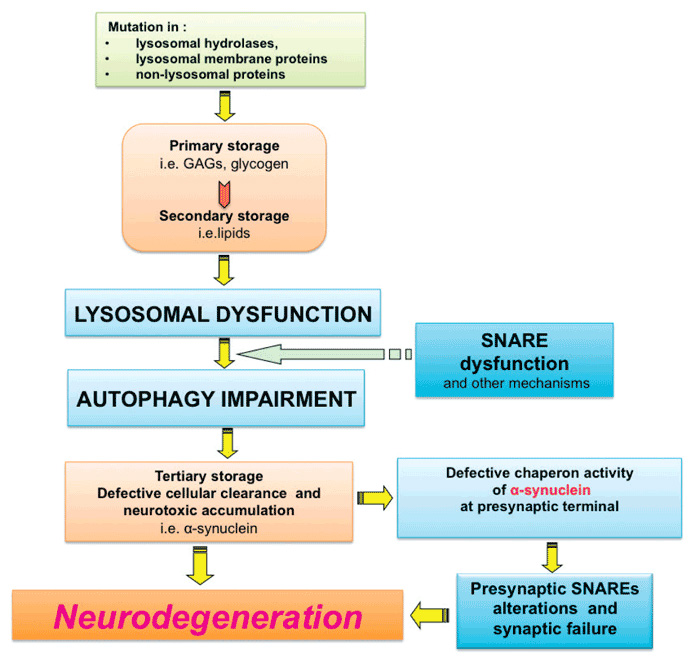
The figure illustrates the main steps determining LSD neuropathogenesis. Mutations in genes, which are important for lysosomal degradation activity result in the lysosomal accumulation of specific un-degraded substrates (Primary storage). This leads to the accumulation of other lysosomal substrates (Secondary storage) due to the secondary inhibition of lysosomal degradation capacity. Lysosomal dysfunction is associated with autophagy impairment, due to the inefficient fusion between lysosomes and autophagosomes. Defective function of SNARE proteins has also been associated with lysosomal fusion defects. Autophagy stress causes tertiary storage of dysfunctional organelles and neurotoxic molecules (e.g. α-synuclein). α-synuclein accumulation may contribute to neurodegeneration through interacting with presynaptic SNARE proteins leading to synaptic failure through the defective chaperone activity of this protein at the presynaptic nerve terminals.
Lysosomal dysfunction has been associated with neuropathology not only in LSDs but also in the most common forms of neurodegenerative disorders such as Parkinson’s, Huntington’s and Alzheimer’s diseases.9
In Huntington’s disease the aggregate-prone proteins huntingtin (HTT) may affect the efficiency of autophagy by inhibiting cargo recognition by autophagosomes.10
Patients with Alzheimer’s disease carrying mutations in presenilin 1 (PSEN1) showed lysosomal and autophagic dysfunction.11 Lysosome dysfunction in these patients can be explained by two different mechanisms, one involving a defect in the lysosomal acidification machinery and the other a defect in lysosomal Ca+2 homeostasis.12
Another aggregate-prone protein, α-synuclein, forms intra-neuronal inclusions called Lewy bodies in Parkinson’s disease (PD). A significant number of patients with Parkinson’s disease are heterozygous for mutations in the gene encoding the lysosomal enzyme β-glucocerebrosidase (GBA) – which when present as homozygous mutations cause Gaucher’s disease, a neurodegenerative LSD. It has been shown that lower levels of GBA lead to an increased accumulation of glucosylceramide in the lysosome, that in turn accelerates the synthesis of soluble α-synuclein oligomers that eventually are converted into amyloid fibrils.13 Furthermore, the accumulation of α-synuclein also blocks the trafficking of newly synthesised GBA to the lysosome and thus further amplifies glucosylceramide accumulation.14 All of these findings indicate that if α-synuclein can somehow be cleared, alpha-synucleinopathies can be prevented or even reversed. An interesting example has emerged recently from the work of McNeil and colleagues30 in which they demonstrated that a commercial drug called “ambroxol”, is able to increase the amount of the enzyme GBA. These results strongly suggest that ambroxol hydrochloride should be further investigated as a potential treatment for PD and α-synuclein -dependent neuropathologies such as LSDs.
In addition, mutations in ATPase type 13A2 (ATP13A2), a component of the lysosomal acidification machinery, has also been found in patients with hereditary parkinsonism and are associated with lysosomal dysfunction, defective clearance of autophagosomes and accumulation of α-synuclein.15 Furthermore, mutations in the genes encoding PINK (PTEN-induced putative kinase) and PARKIN (Parkinson’s disease protein) are associated with the defective clearance of mitochondria, leading to Parkinson’s disease.16 Finally there are also some Parkinson’s disease patients with VPS35 (vacuolar protein sorting 35) mutations, which encodes for an endosomal protein involved in the retrograde transport between endosomes and the trans-Golgi network.17
Neurodegenerative mechanisms in LSDs
The prominent pathological hallmark of LSDs is a severe neurodegeneration, which is connected to lysosome dysfunction. A variety of lysosomal-dependent pathogenic cascades are activated in LSDs, such as impaired autophagic flux, altered calcium homeostasis, oxidative stress, inflammation, altered lipid trafficking, endoplasmic reticulum stress and autoimmune responses. The mechanisms linking these pathogenic cascades to lysosomal dysfunction are now being better understood.18,19,20 The impairment of autophagy plays an important role in the pathogenesis of LSDs.21 In neurons the failure of autophagy results in secondary accumulation of toxic substrates, aggregate-prone proteins and damaged organelles. Toxic storage triggers neurodegenerative processes since it is a critical determinant of cell death in post-mitotic cells such as neurons. Indeed, intact autophagic pathways are crucial to maintain normal neuronal function22 and any decline of their degradation capability contributes to the pathogenesis not only of LSDs but also to age-related neurodegenerative disorders.23 A critical mechanism underlying lysosomal dysfunction and autophagic impairment in LSDs involves soluble NSF attachment receptor (SNARE) proteins. SNAREs are a group of proteins that are responsible for mediating membrane fusion processes in cells. By studying two neurodegenerative LSDs, the mucopolysaccharidosis type IIIA (MPS-IIIA) and Multiple Sulfatase Deficiency (MSD), it has been demonstrated that cholesterol accumulation in lysosomal membranes impairs SNARE function thus leading to defective fusion of lysosomes with target membranes including autophagosomes.24 Furthermore SNARE impairment has been identified as an important mechanism underlying lysosomal dysfunction and autophagic defects in NPC-125 as well.
An additional pathogenic process that has recently been put forward is based on the fact that α-synuclein accumulation has been found in a number of LSDs (ref: Chachar et al Movement Disorders, 2011). Moreover, patients heterozygous for GBA mutations can develop PD with its alpha synuclein Lewy bodies. Now exactly how α-synuclein accumulation and aggregation mediates neurotoxicity remains still unclear but recently, α-synuclein has been identified as a key chaperone protein assisting synaptic vesicle recycling and transmission at presynaptic terminals. Specifically, α-synuclein ensures the proper function of synaptic SNARE proteins, which represent the key component of the neuronal cellular fusion machinery at nerve terminals.31,32 This evidence raises the intriguing hypothesis that lysosomal-dependent α-synuclein accumulation found in LSDs may contribute to trigger neurodegeneration in these pathologies by causing α-synuclein chaperone deficits and presynaptic failure. This may represent a new mechanistic link between lysosomal dysfunction and neuronal degeneration in LSDs (Figure 1).
In conclusion, understanding how the failure of the lysosomal system impacts on neuropathology in LSDs may uncover new pathogenic cascades and through this new therapeutic agents. Moreover, by studying neuropathogenic mechanisms in LSDs this may help clarify not only the cell biology of lysosomal diseases but the much more common age-related neurodegenerative disorders.
References
- K, S. Lysosomal disease. In Greenfield’s Neuropathology, Graham DI, Lantos PL (eds) 653–735(2002).
- Ballabio, A. & Gieselmann, V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta 1793, 684-696 (2009).
- Settembre C, Fraldi A, Medina DL & Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013;14:283-96.
- Mizushima N. Autophagy: process and function. Genes & development 2007;21:2861-73.
- Andrews NW, Almeida PE & Corrotte M. Damage control: cellular mechanisms of plasma membrane repair. Trends in cell biology (2014).
- Medina DL, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Developmental cell 2011;21:421-30.
- Settembre C, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. The EMBO journal 2012;31:1095-108.
- Cuervo AM & Dice JF. When lysosomes get old. Experimental gerontology 2000;35:119-31.
- Nixon RA, Yang DS & Lee JH. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 2008;4:590-9.
- Jeong H, et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009;137:60-72.
- Lee JH. et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010;141:1146-58.
- Coen K, et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. The Journal of cell biology 2012;198:23-35.
- Sidransky E, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. The New England journal of medicine 2009;361:1651-61.
- Mazzulli JR, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011;146:37-52.
- Ramirez A, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nature genetics 2006;38:1184-91.
- Geisler S, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature cell biology 2010;12:119-31.
- Zimprich A, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. American journal of human genetics 2011;89:168-75.
- Schultz ML, Tecedor L, Chang M & Davidson BL. Clarifying lysosomal storage diseases. Trends in neurosciences 2011;34:401-10.
- Platt FM, Boland B & van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. The Journal of cell biology 2012;199:723-34.
- Vitner EB, Platt FM & Futerman AH. Common and uncommon pathogenic cascades in lysosomal storage diseases. The Journal of biological chemistry 2010;285:20423-7.
- Settembre C, Fraldi A, Rubinsztein DC & Ballabio A. Lysosomal storage diseases as disorders of autophagy. Autophagy 2008;4:113-14.
- Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006;441:880-4.
- Harris H & Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nature reviews. Neurology 2012;8:108-17.
- Fraldi A, et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. The EMBO journal 2010;29:3607-20.
- Sarkar S, et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell reports 2013;5:1302-15.
- Spillantini MG & Goedert M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Annals of the New York Academy of Sciences 2000;920:16-27.
- Bendor JT, Logan TP & Edwards RH. The function of alpha-synuclein. Neuron 2013;79:1044-66.
- Bourdenx M, Bezard E & Dehay B. Lysosomes and alpha-synuclein form a dangerous duet leading to neuronal cell death. Frontiers in neuroanatomy 2014;8:83.
- Xu YH, et al. Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Molecular genetics and metabolism 2011;102:436-47.
- McNeill A, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain: a journal of neurology 2014;137:1481-95.
- Burgoyne RD & Morgan A. Chaperoning the SNAREs: a role in preventing neurodegeneration? Nature cell biology 2011;13:8-9.
- Burre J, et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010;329:1663-7.