

Neurofibromatosis is the umbrella term for three distinct genetic disorders (NF1, NF2 and Schwannomatosis) that share the hall mark of tumour growth on the tissues surrounding nerves. It is a misnomer in Neurofibromatosis type 2 (NF2), as the predominant tumours are schwannomas and meningiomas.
Epidemiology
The birth incidence of NF2 is 1 in 33,000 and prevalence, 1 in 60,000.1 There is no predilection for sex, race or ethnic origin. Approximately half of the cases are familial and half are sporadic,2 due to a spontaneous mutation in the NF2 gene.
Diagnostic criteria
The first case report of neurofibromatosis appeared in the early 19th century. It gradually became clear that there were two clinically distinct entities, ‘peripheral neurofibromatosis’, or von Recklinghausen’s disease and ‘central neurofibromatosis’. It wasn’t until 1987 that these were described as NF1 and NF2 respectively and diagnostic criteria were established.3 These have subsequently been reviewed and updated in 1990,4 1997 (Box 1)5 and most recently in 2011 (Box 2).6
The diagnosis of NF2 remains clinical, although in most cases can be confirmed with molecular testing. All of the tissues may not be affected and all the potential genetic mutations have not been identified for a reliable genetic test. The diagnosis can therefore take years as many of the features are age dependant. The hallmark is bilateral vestibular schwannomas. The numerous changes to the diagnostic criteria reflect the difficulty in establishing a confident diagnosis as early as possible in those who do not (yet) have bilateral vestibular schwannomas, or a positive family history. The most recent (Baser) criteria are listed in Box 2. They incorporate mutation testing for those with ‘probable’ NF2 and have a diagnostic sensitivity of 79% and a specificity of 100%.6
Genetics
The NF2 gene is a tumour suppressor gene located on the long arm of Chromosome 22 (22q12.2). It was discovered independently by two teams who named the encoded protein merlin7 and schwannomin8 respectively. This is expressed in many different cells including neurons, schwann cells, fibroblasts and some meningiomas. There are two isoforms of the protein depending on the location.2,9 Only one of them can acquire the conformation to allow tumour suppressor activity.10 The name merlin (moesin, ezin and radixin-like protein) reflects the close similarity to a group of proteins including moesin, ezrin, radixin, talin and erythrocyte protein 4.1 which regulate interactions with the cell cytoskeleton, determining cell shape and influencing cell division and interactions with other cells and the extracellular matrix. Many mutations have been located throughout the NF2 gene. Most (90%) have the effect of truncating the transcribed protein.9 Both copies of the NF2 gene are mutated within tissue of NF2 related vestibular schwannomas and meningiomas and 30-70% of sporadic meningiomas i.e. tumour formation is initiated when there is a ‘second hit’ to the gene, supporting the theory that merlin is a tumour suppressor.
Familial NF2 is inherited in an autosomal dominant manner with almost 100% penetrance. It accounts for 50% of NF2.2 Sporadic cases may be prezygotic, ie the mutation is present in one of the gametes and therefore uniformly distributed through all of the cell lines, or post zygotic. The stage at which post zygotic mutations occur in development and cell differentiation, determines the distribution of the mutation and thus the distribution and severity of the clinical manifestations. 25-33% of those with sporadic NF2 have somatic mosaicism2,11,12 where the genetic defect is restricted to a group or population of cells. NF2 can therefore be difficult to diagnose in such individuals as genetic testing may be normal in unaffected tissues and they may not have the pathognomonic clinical feature of NF2, bilateral VS.2 When mosaicism levels are greater than 10% the mutation will be present in lymphocyte DNA2 and therefore detectable on blood tests. Tumour material is required to detect the mutation when the mosaicism is less.
Genetic testing can be used to confirm a diagnosis of NF2 and/or to identify the mutation which is invaluable for screening relatives and offspring. The tests available are not 100% sensitive as the gene is large and there are so many potential mutations. Gene sequence analysis or mutation analysis, combined with deletion / duplication analysis can achieve a detection rate of more than 90% of pathogenic inherited NF2 mutations on blood testing.2 The detection rate is reduced to 64% in de novo cases as around one third of these cases have low levels of mosaicism that are not detectable in lymphocyte DNA6,11,12 and overall the mutations are less easy to detect as they include a smaller percentage of large deletions.2
Where a causal mutation has been identified within a family, other members can confidently be tested to confirm or exclude the mutation. The risk of transmission is very low in those with somatic mosaicism, when the mutation is only detectable in the tumour.11
The Gene Tests laboratory directory (http://www.genetests.org) lists the testing available from genetics laboratories world wide. The genetics laboratory in Manchester (http://www.mangen.co.uk/) is the only one in the UK to offer NF2 testing. Any tumour samples should be sent fresh or frozen to allow DNA sampling and accompanied by a lymphocyte DNA sample.
Clinical presentation / Natural history
The clinical manifestations and the severity of NF2 vary enormously. There is a strong genotype-phenotype correlation. In inherited NF2 the severity of the condition and its progression is similar within successive generations of the same family9 and the type of mutation appears to be an important determinant. Nonsense, frameshifting and truncating mutations are associated with more severe disease.2 Those with mutations that cannot be identified tend to have milder disease.11 With sporadic NF2 it is more difficult to predict the course of the condition; however those with mosaicism tend to have a milder form. Two forms of NF2 have been described, to denote the aggressiveness of the disease. The Wishart type is more aggressive, the Gardner type is less aggressive, presenting in older patients. Overall the average age of onset is 18-24 years.2
Whatever the form of NF2 it is impossible to predict the location and types of tumours that will develop. Manifestations of NF2 include schwannomas, meningiomas, ependymomas, rarely, low grade astrocytomas and eye conditions including retinal hamartoma.
Schwannomas
Almost all adults with NF2 will develop bilateral vestibular schwannomas (VS) (Figure 1), which present in a similar fashion to sporadic VS (see Ramnarine D, Whitfield P. Management of patients with vestibular schwannomas. ACNR 2005;(4):24-5). However NF2 related VS tend to be more aggressive in terms of rate of growth, recurrence and involvement of the facial nerve. Most affected individuals have developed bilateral VS by 30 years of age.2 Also, given the propensity of NF2 patients to develop schwannomas and meningiomas, one must consider whether what on first impression appears to be a VS may be a schwannoma arising from another cranial nerve such as the cochlear, facial or trigeminal, or a CP angle meningioma. All of the other cranial nerves are potential sites for schwannomas, though the trigeminal is the most common. Trigeminal schwannomas can extend above and below the tentorium and involve the cavernous sinus.
{kind=link}
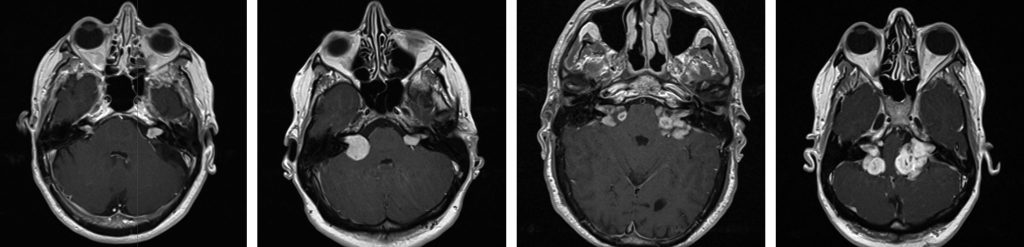
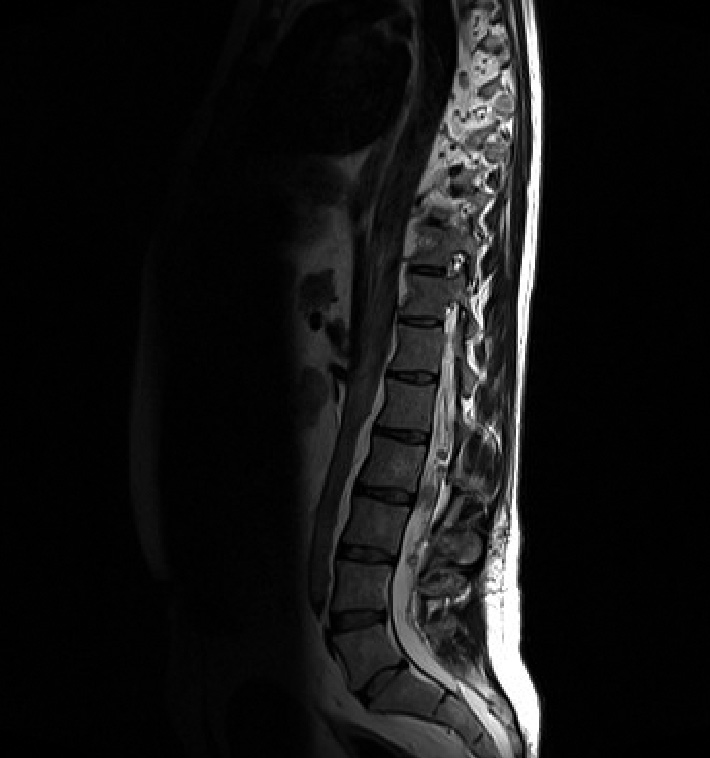
Spinal schwannomas (Figure 2)
The most common type of spinal tumour in NF2. They are often multiple, dotted throughout the cauda equina and those in the cervicothoracic region are often indistinguishable from the neurofibromas of NF1. They originate on the dorsal root and adopt a dumb bell shape as they extend through the lateral and medial intervertebral foramen. NF2 patients can also develop schwannomas of the peripheral nerves and dermis.
Meningiomas
Approximately 50-75% of NF2 patients develop meningiomas, most commonly in supratentorial locations.9 These present in a similar way to sporadic meningiomas. They are usually of the fibroblastic type.2
Ependymomas and low grade Astrocytomas
These intrinsic tumours tend to occur in the spinal cord and brain respectively. They are rarely the predominant symptomatic lesion in NF2.
Eye conditions
These are common and tend to develop early in the course of NF2. One third of individuals develop reduced visual acuity. Problems include early cataracts, an important diagnostic feature, retinal hamartomas, epiretinal membrane,10 strabismus and amblyopia, orbital meningiomas and corneal damage following loss of facial and trigeminal nerve function.
Neuropathy
NF2 is associated with a mononeuropathy of childhood and a polyneuropathy of adulthood. Children may present with a facial palsy, unrelated to a tumour mass that partially recovers, a third nerve palsy, a hand or foot drop2 or a polio like illness with wasting of the lower limb muscles.13 Approximately 40% of adults will have evidence of polyneuropathy on nerve conduction studies and 3-10% will be symptomatic.13
{kind=link}
Skin
Seventy percent of NF2 patients develop skin lesions which include skin plaques, subcutaneous tumours and intradermal tumours. The cutaneous features of NF2 are far more discreet than those of NF1. Intracutaneous tumours/skin plaques tend to be raised, slightly pigmented plaques which may have associated hair.10
Congenital Dysplasias
Increasing numbers of children are being screened and diagnosed with NF2. With the improvement in the resolution of MR Imaging, we are increasingly recognising congenital glial and vascular abnormalities in children and young adults with NF2.
Assessment and Investigations
NF2 patients may harbour many or all of these pathologies at various locations. It is therefore very important to perform a thorough history and neurological and radiological assessment to establish which are symptomatic and whether there has been any deterioration. Patients with an established diagnosis, or a strong suspicion of NF2 are now managed in specialist multidisciplinary clinics where they are seen by neurologists, geneticists, an ophthalmologist and skull base surgeons with the support of neuroradiologists, audiologists and specialist nurses. However, it is not unheard of for a patient with a VS or meningioma, under follow up in a general neurosurgical clinic, to develop further symptoms or lesions on surveillance scanning, suggestive of NF2.
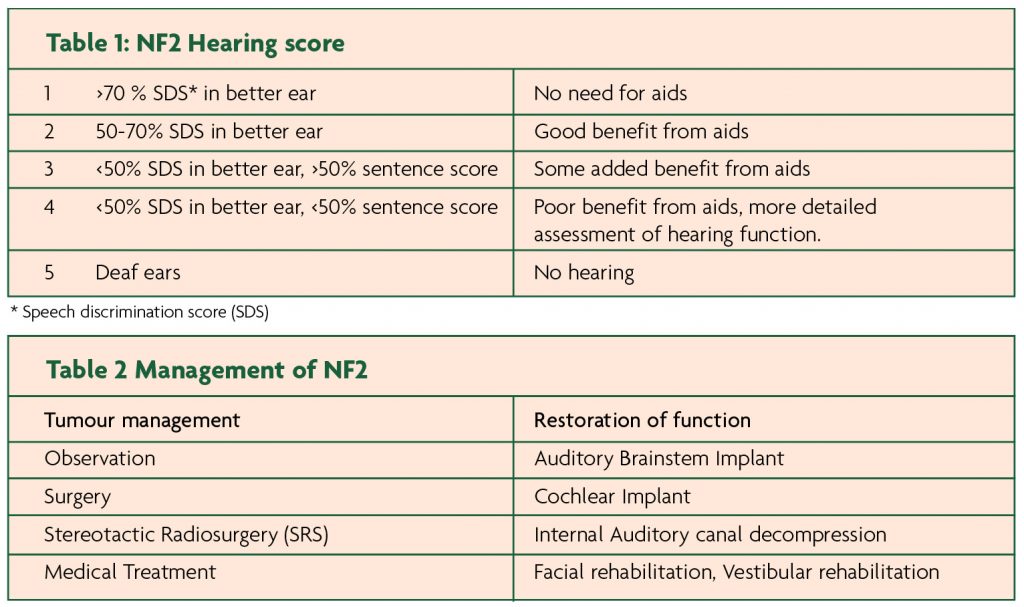
During the history pay particular attention to hearing; can they use the telephone? Do they use any aids? Do they have any balance disturbance, which many quality of life studies report to be more disabling than hearing loss. The NF2 service in the UK uses the NF2 hearing score (Table 1). A very functional categorisation of hearing, using the speech discrimination score. Also ask about symptoms suggestive of other cranial nerve neuropathies eg diplopia, dysphagia, visual disturbances, peripheral numbness or weakness that might be secondary to peripheral neuropathy or spinal disease, seizures and a detailed family history if they are a new patient.
Management
NF2 is a condition of multiple slow growing tumours which rarely turn malignant. The main ethos of management is therefore preservation of function, rather than cure. Management options can be classified into those directed at tumour treatment and those directed at restoration of function (Table 2).
This generally involves a strategy of watchful waiting where tumours are treated when they become symptomatic or are clearly growing radiologically. There is still debate. Some advocate an early, anticipatory approach to stop tumours becoming symptomatic eg the treatment of small VS with stereotactic radiosurgery (SRS) or hearing preservation surgery.14
The first priority is to evaluate the disease load and symptoms with full brain and spinal MRI, neurological examination, ophthalmological examination, full hearing assessment including brainstem auditory evoked responses (BAERs), cutaneous examination and genetics consultation. Unless symptoms progress, patients should undergo regular annual neurological, ophthalmology and audiology assessment as well as brain MRI. Spinal MRIs are generally performed every four to five years unless patients develop worrying symptoms or lesions.
Vestibular schwannomas
NF2 related VS are more difficult to treat than their sporadic counterparts. This is because they are often large by the time they are treated and they tend to be more aggressive in nature. Therefore, whatever the treatment modality, the risk of recurrence, facial nerve injury and other complications is higher.
SRS: It is widely accepted that Stereotactic radiosurgery is less effective in NF2 related VS. Relative growth ratios with contralateral controls indicate that SRS does have a beneficial effect in slowing tumour growth.15 Approximately 50% are controlled at eight years.15 Tumour volume is the main determinant of outcome and that coupled with their propensity to grow, restricts the size of tumour suitable for treatment. The Sheffield group suggest a volume limit of 10cm3.16 There is a 5% risk of facial palsy and whilst there is great individual variation in the rate of hearing loss, overall SRS does not appear to slow the rate of deterioration.15 Many have concerns about the risk of malignancy, delivering radiation to a tumour prone condition.17 In contrast, others advocate fractionated stereotactic radiotherapy in an attempt to preserve hearing and facial nerve function.
Surgery: Surgery remains the primary treatment for NF2 related vestibular schwannomas. Most are large, in which case, hearing preservation surgery is not possible. The timing of surgery is a balance between hearing, facial nerve function and brainstem compression. When there is no functional hearing, the threshold for surgery is lower, by which time the tumour may have reached a considerable size, increasing the risk of facial nerve injury and other surgical complications.
Some advocate an early surgical approach to small intracanalicular VS by a middle fossa approach with hearing preservation rates of up to 50%.14
Hearing Preservation and Augmentation
All NF2 patients should be referred to an audiologist for regular hearing assessments and training in anticipation of losing their hearing. Hearing and communication may be optimised by hearing aids initially. Lip reading, sign language and cochlear or auditory brainstem implants are options for those who have completely lost their hearing.
Of the surgical implants, cochlear implants provide a better quality of auditory input however they require an intact cochlear nerve. The scenario where this might occur in NF2 related VS is following attempted hearing preservation surgery or where there is a stable vestibular schwannoma with no hearing loss. Where there is early hearing loss secondary to a small intracanalicular VS, one option is to decompress the internal auditory canal, via a middle fossa approach.18
Auditory Brain Stem Implants (ABI): ABIs are used to treat total deafness when there has been permanent damage to the cochlear nerves and hearing can therefore not be improved by other (more effective) means such as hearing aids or cochlear implant. Electrodes are placed on the cochlear nucleus at the auditory prominence, through the Foramen of Luschka in the lateral recess of the 4th ventricle, guided by electrophysiology recordings of the brainstem auditory evoked responses. This is connected to an external receiver and sound processor which convert sounds into electrical signals. The auditory sensations that are gained are a perception of environmental sound which can be a great aid to lip reading. Excellent speech perception is possible. ABIs are often placed immediately following removal of vestibular schwannomas in the same surgical sitting. In patients with NF2 and bilateral vestibular schwannomas, ‘sleeper’ ABIs are increasingly being placed with the removal of the first VS in anticipation of complete hearing loss as the opposite VS grows or is treated. There is some growing evidence that patients can derive greater benefit from their ABI if they have the opportunity to practice using it particularly whilst they have some residual normal hearing (personal communication). Overall, the outcomes with ABIs are extremely variable ranging from non users to excellent hearing function. There are no clear predictors of outcome.19 The external receiver contains a magnet that risks getting dislodged during MRI scanning. This is not an absolute contraindication to MRI imaging but special precautions need to be taken. Our local protocol includes head wrapping a protective guard over the device.
Medical treatment
There is an ongoing search for an effective medical treatment for NF2 related tumours. Phase 2 trials are under way for Lapatinib, a tyrosine kinase inhibitor at EGFR receptors, Everolimus an immunosuppressant and Bevacizumab/Avastin. The latter is a monoclonal antibody that inhibits vascular endothelial growth factor and therefore tumour angiogenesis. It is currently approved for off label use in England under strict criteria including active schwannoma growth of at least 4mm or 60% volume in 12 months, in the context of severe multi tumour disease. It is suggested in situations where the tumour load is so high that targeted surgery would not be possible or beneficial or where there is loss of hearing secondary to growth in the only hearing ear, in an effort to re establish and preserve hearing for as long as possible. The initial observations are that Avastin is very effective at halting tumour growth and is therefore being increasingly used with rapidly growing tumours. The main disadvantages of Avastin therapy are that it poses a contraindication to surgery which is delayed until three months after stopping treatment and the benefits tend to last as long as the treatment, such that there is a risk of rebound on stopping. It also does not have any real efficacy against meningiomas.
Facial reanimation
NF2 patients may develop facial paresis secondary to mononeuropathy, tumour growth or following surgery. Several options are available to reduce this disfiguring palsy and to protect the cornea, including, botulinum injections, gold eyelid weights and surgery. Restoration of the continuity of the facial nerve by cable graft following the removal of a large VS is not possible as the proximal stump will be very short and unidentifiable with post operative changes in the internal auditory canal. In such circumstances cross over techniques such as end to end or end to side hypoglossal facial anastomosis can be employed. The strategy for facial reanimation depends on the grade of facial weakness, the duration of the facial palsy, the status of the underlying tumour and other NF2 related tumours.
Children with a family history of NF2
If the mutation has been identified within the family then the diagnosis / mutation can easily be confirmed or excluded with direct DNA testing. Screening with MRI usually begins at 10-12 years in those known to carry a disease causing mutation or those at risk of inheriting NF2 where the mutation is unknown. However the timing is guided by family history and should be earlier if the family history is of early onset NF2. If the diagnosis cannot be excluded annual MRIs are continued until at least the fourth decade of life.2
In the UK, NF2 is one of the rare conditions whose services are commissioned at a National level. The rationale is that concentrating the management of this complex condition in a few (4) centres will increase the experience, quality and ‘uniformity’ of the overall service.
References
- Evans DG, Howard E, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am. J. Med. Genet. A. 2010 Feb;152A(2):327–32.
- Evans DG, Pagon RA TDB. Neurofibromatosis 2 [Internet]. GeneReviews Editors: Pagon RA, Bird TD, Dolan CR, Stephens K. 1998 [cited 2011 Sep 14]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1201/?report=printable
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988 May;45(5):575–8.
- Mulvihill JJ, Parry DM, et al. NIH conference. Neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis). An update. Ann. Intern. Med. 1990 Jul 1;113(1):39–52.
- Gutmann DH, Aylsworth A, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997 Jul 2;278(1):51–7.
- Baser ME, Friedman JM, et al. Empirical development of improved diagnostic criteria for neurofibromatosis 2. Genet. Med. 2011 Jun;13(6):576–81.
- Trofatter JA, MacCollin MM, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993 Nov 19;75(4):826.
- Rouleau GA, Merel P, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993 Jun 10;363(6429):515–21.
- Korf BR, Rubenstein AE. Neurofibromatosis: A Handbook for Patients, Families and Health Care Professionals. 2nd ed. Thieme Medical Publishers; 2005.
- Asthagiri AR, Parry DM, et al. Neurofibromatosis type 2. Lancet. 2009 Jun 6;373(9679):1974–86.
- Moyhuddin A, Baser ME, et al. Somatic mosaicism in neurofibromatosis 2: prevalence and risk of disease transmission to offspring. J. Med. Genet. 2003 Jun;40(6):459–63.
- Kluwe L, Mautner V, et al. Molecular study of frequency of mosaicism in neurofibromatosis 2 patients with bilateral vestibular schwannomas. J. Med. Genet. 2003 Feb;40(2):109–14.
- Evans GR, Lloyd SKW, Ramsden RT. Neurofibromatosis type 2. Adv. Otorhinolaryngol. 2011;70:91–8.
- Slattery WH 3rd, Fisher LM, et al. Hearing preservation surgery for neurofibromatosis Type 2-related vestibular schwannoma in pediatric patients. J. Neurosurg. 2007 Apr;106(4 Suppl):255–60.
- Rowe J, Radatz M, et al. Clinical experience with gamma knife stereotactic radiosurgery in the management of vestibular schwannomas secondary to type 2 neurofibromatosis. J Neurol Neurosurg Psychiatry. 2003 Sep;74(9):1288–93.
- Rowe JG, Radatz M, et al. Stereotactic radiosurgery for type 2 neurofibromatosis acoustic neuromas: patient selection and tumour size. Stereotact Funct Neurosurg. 2002;79(2):107–16.
- Carlson ML, Babovic-Vuksanovic D, et al. Radiation-induced rhabdomyosarcoma of the brainstem in a patient with neurofibromatosis type 2. J. Neurosurg. 2010 Jan;112(1):81–7.
- Slattery WH, Hoa M, et al. Middle Fossa Decompression for Hearing Preservation. Otology & Neurotology. 2011 Aug;32(6):1017–24.
- Sanna M, Di Lella F, et al. Auditory brainstem implants in NF2 patients: results and review of the literature. Otol. Neurotol. 2012 Feb;33(2):154–64.