



Abstract
Over the past 13 years neuromyelitis optica spectrum disorder (NMOSD) has emerged as a discrete form of demyelinating disease of the central nervous system in which antibodies against a water channel found in high concentration on astrocytes are frequently found. Following the discovery of this pathogenic antibody the phenotype of this condition, previously known as Devic’s disease, has broadened from one of monophasic or recurrent, optic neuritis and transverse myelitis, to include area postrema lesions, hypothalamic lesions and a fulminant encephalopathic presentation. Clues to the diagnosis include clinical presentations related to the above locations of pathology and imaging changes including longitudinally extensive spinal cord lesions, extensive optic nerve lesions, particularly lesions posterior to or involving the chiasm, and reactive cerebrospinal fluid. Early identification of this disorder is important as the prognosis without treatment is poor and generally worse than is seen in multiple sclerosis (MS). In addition, the approach to treatment (immunosuppression and anti-B-cell therapy) is different to MS and there is a concern that some disease modifying therapies that are helpful in MS may be harmful in NMOSD.
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is an antibody-mediated, inflammatory disease of the central nervous system primarily affecting the optic nerves, spinal cord and periependymal regions of the cerebral hemispheres and brainstem.1 Following the initial descriptions of what became known as Devic’s disease, monophasic, sequential and relapsing forms of a distinct pathological entity with a predilection for the optic nerves and spinal cord had been recognised for over a century.2 The discovery of antibodies against the aquaporin 4 (AQP4) water channel in 2004 has enabled a broadening of the phenotype of this astrocytopathy to include a core group of six clinical syndromes.3
Pathogenesis
Antibodies to AQP4 play a key role in the pathogenesis of NMOSD.4 AQP4 is a water channel that is predominantly expressed on the cell membrane of astrocytic end-feet, forming part of the blood–brain barrier. Predilection for particular regions of the CNS in NMOSD is related to higher expression of AQP4 in the optic nerves and spinal cord and a lack of tight junctions between endothelial cells forming a permeable blood-brain barrier in these areas.1 Binding of antibody downregulates AQP4 and causes astrocytic injury through activation of the classical complement pathway. Antibody-complement complex formation results in chemotaxis of T and B lymphocytes, macrophages, neutrophils and eosinophils, principally through activation of NFκB.4 Demyelination and oligodendrocyte injury occur as a secondary effect of this immune response.1 This pathological picture is distinct from and more destructive than that seen in MS. The main pathological features of NMOSD are illustrated in Figure 1.
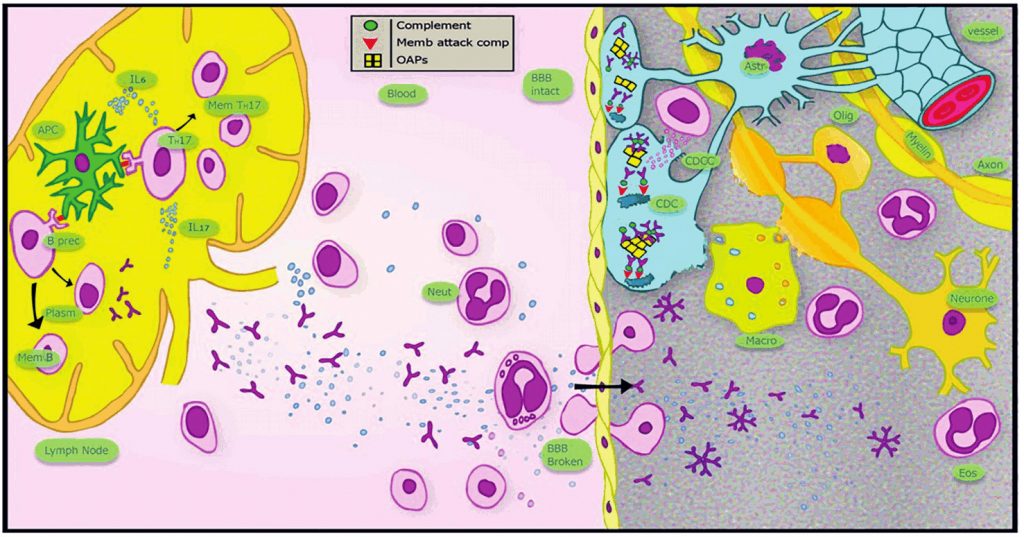
Epidemiology
The incidence of NMOSD ranges from 0.053 to 0.4 per 100,000 individuals with prevalence rates from 0.52 to 4.4 per 100,000.5 NMOSD is more prevalent among non-Caucasians and is relatively rare in childhood.5 Onset is more evenly spread across the adult age range than is seen in MS and the mean age at onset is higher (40-45 vs. 30-35 years).5
Clinical features
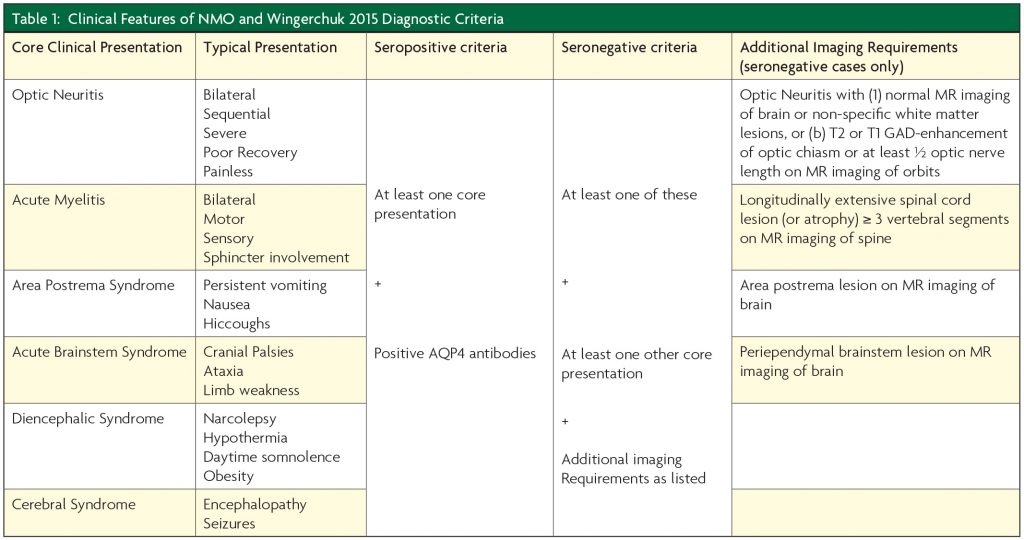
The common presenting features of NMOSD can be attributed to lesions of the optic nerve, spinal cord, area postrema, periependymal regions of the third ventricle and hypothalamus, brainstem and cerebral hemispheres.3 The typical clinical presentations of NMOSD are given in Table 1. The key to recognising NMOSD as the diagnosis in patients presenting with demyelinating disease of the central nervous system is the early identification of the listed clinical and radiological features for each type of presentation and having a low threshold for requesting AQP4 antibody testing.
Clinical course and prognosis
The majority of NMOSD patients have a relapsing disease course and the presence of AQP4 anti- bodies is predictive of a relapsing course in limited forms of the disease (e.g. monophasic transverse myelitis). Unlike MS, a secondary progressive phase is uncommon in NMOSD.6 However, significant permanent disability resulting from acute attacks due to frequently necrotic pathology is common in NMOSD.3 As a consequence the prognosis without treatment in NMO is generally worse than for MS.
Associated autoimmune disease
Around 20 – 30% of NMOSD patients have a co-existing autoimmune disease and autoantibodies other than anti-AQP4 can be detected in up to 40%.7 Case reports of NMOSD being associated with cancer suggest that the disease may occasionally occur as a paraneoplastic phenomenon.7
Diagnosis and investigations
In 2015 The International Panel for NMO Diagnosis unified the concept of NMO and NMOSD and developed the revised diagnostic criteria based on AQP4-IgG status in which more strict clinical criteria, with additional neuroimaging findings are required for diagnosis of NMOSD when AQP4 antibodies are absent or where serologic testing is unavailable.3 The key elements of these criteria are given in Table 1.
Imaging
The most distinct manifestation of NMOSD is a longitudinally extensive spinal cord lesion, defined as a lesion that spans over three or more contiguous vertebral segments and predominantly involves central grey matter.8 Non-specific, white matter, dots and patches of T2/FLAIR hyperintensity are the most common findings on MR images of the brain in NMOSD.8 Lesions in the dorsal brainstem adjacent to the fourth ventricle including the area postrema and the nucleus tractus solitarius are more specific for NMOSD. Other typical brain lesions include bridging lesions of the splenium, and periependymal lesions of the ventricles and aqueduct. Lesions of the corpus collosum tend to be more heterogeneous than those seen in MS. Lesions of the corticospinal tracts may also be seen.8 Increased signal within the optic nerve may be detected with fat suppressed T2-weighted orbital MRI sequences, typically with gadolinium enhancement seen on T1-weighted sequences. Bilateral optic nerve involvement, posterior nerve predominance (especially with extension into the optic chiasm), or extensive lesions of the optic nerve (more than half of its length) are all suggestive of NMOSD.3 Typical MR imaging appearances of NMOSD are shown in Figure 2.
Serological testing
Anti-AQP4 antibodies
Antibodies against AQP4 are detected in a high proportion of patients with NMOSD (70 – 80%).9 A number of different assays are available, including immunofluorescent histological techniques, enzyme-linked immunosorbent assay (ELISA), dried cell-based assays and live cell-based assays with and without uorescence assisted cell sorting. Evidence suggests that the live-cell based assays are the most sensitive.9 All appear to be highly specific (97% or higher) with the exception of ELISA methods. ELISA can be useful in providing a ready measure of the antibody titre.9 Many laboratories use a combination of techniques to improve sensitivity and specificity.
Anti-MOG antibodies
The finding of antibodies targeted against myelin oligodendrocyte glycoprotein (MOG) in a small proportion of AQP4 seronegative NMOSD has raised the question of whether or not these patients form a subgroup of NMOSD. However, an emerging phenotype of ADEM in childhood and recurrent optic neuritis or classical Devic’s presentation (simultaneous optic neuritis and acute myelitis) in adolescence and adults is defining this condition as a separate demyelinating disease which has been coined ‘anti-MOG related demyelinating disease’.10 A typical MR imaging feature of this condition is a longitudinally extensive spinal cord lesion extending all the way down to the conus. The optic neuritis in this condition is typically highly steroid sensitive.
Other Investigations
Elevation of cerebrospinal fluid (CSF) protein and CSF pleocytosis are more commonly seen during acute attacks of NMOSD and are of some assistance in distinguishing NMOSD from MS.11 In MS, a CSF white cell count greater than 10 per ml is rare. Oligoclonal bands in the CSF are seen less often than in MS, but can be present in up to 20% of patients with NMOSD.11 Somatosensory evoked potentials, brainstem acoustic evoked potentials and visual evoked potentials are frequently abnormal when symptomatic lesions are tested in NMOSD, but unlike MS asymptomatic abnormalities are rare12 and there is no evidence for peripheral motor and sensory nerve conduction abnormalities in NMOSD.13
Optical coherence tomography in NMOSD shows significantly greater retinal nerve fibre layer thinning than is typically seen in MS, reflecting a more severe axonal injury.14 It has been proposed that this may be a useful indicator for potential NMOSD cases, but these changes may take several weeks or even months to become established following an acute attack of optic neuritis.
Treatment
Acute exacerbations should be treated promptly with high-dose intravenous methylprednisolone for 3-5 days. Most authors recommend a subsequent taper of oral prednisolone over 2-3 months to prevent relapse, particularly when the level of deficit at presentation is high. The period of oral steroids can be adjusted in the light of other adjunctive therapies. Where immediate improvement is not seen, a low threshold for the early implementation of plasma exchange is recommended. Indeed, many would advocate immediate plasma exchange when initial neurological impairment is severe (e.g. paraplegia), in order to optimise recovery.11
For the prevention of relapses azathioprine, mycophenolate mofetil and rituximab are recommended. There are no randomised, placebo controlled or head-to-head comparison trials of these agents in NMOSD, but rituximab may have greater efficacy.11 Mycophenolate mofetil has fewer and milder adverse events compared with azathioprine with similar efficacy.15
Where doubt exists regarding the diagnosis of NMOSD or MS (e.g. clinical or radiological features suggestive of NMOSD in a seronegative patient with MR imaging of brain that is not typical of MS) then treatment with β-interferon, fingolimod and natalizumab should be avoided as there is some evidence to suggest that these therapies may have a negative outcome in NMOSD.16 Glatiramer acetate does not have this drawback and there is some evidence to suggest it may be helpful in NMOSD and anti-B-cell therapies in the form of rituxumab or ocrelizumab have evidence of efficacy in both MS and NMOSD.
Conclusions
NMOSD represents a distinct clinical and pathological entity which has some clinical overlap with MS, optic neuritis and transverse myelitis, but can now be regarded as a unique astrocytopathy. The distribution of AQP4 and the activation of complement explains both the clinical phenotypic expression of the disease and its severity. A high degree of clinical suspicion with any clinical or MR imaging features of NMOSD is crucial for early diagnosis. Aggressive treatment of acute relapses and early adoption of long term preventive therapies are key to minimising the long term adverse outcomes that have been a hall mark of this condition in the past.
References
- Lucchinetti CF, Guo Y, Popescu BF, et al. The pathology of an autoimmune astrocy- topathy: lessons learned from neuromyelitis optica. Brain Pathol 2014;24(1):83-97.
- Wingerchuk DM, Hogancamp WF, O’Brien PC, et al. The clinical course of neuro-myelitis optica (Devic’s syndrome). Neurology 1999;53(5):1107-14.
- Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85(2):177-89.
- Bukhari W, Barnett MH, Prain K, et al. Molecular pathogenesis of neuromyelitis optica. Int J Mol Sci 2012;13(10):12970-93.
- Pereira WL, Reiche EM, Kallaur AP, et al. Epidemiological, clinical, and immunological characteristics of neuromyelitis optica: A review. J Neurol Sci 2015;355(1- 2):7-17.
- Wingerchuk DM, Pittock SJ, Lucchinetti CF, et al. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 2007;68(8):603-5.
- Iyer A, Elsone L, Appleton R, et al. A review of the current literature and a guide to the early diagnosis of autoimmune disorders associated with neuromyelitis optica. Autoimmunity 2014;47(3):154-61.
- Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology 2015;84(11):1165-73.
- Ruiz-Gaviria R, Baracaldo I, Castaneda C, et al. Specificity and sensitivity of aqua- porin 4 antibody detection tests in patients with neuromyelitis optica: A meta-analysis. Multiple sclerosis and related disorders 2015;4(4):345-9.
- Ramanathan S, Dale RC, Brilot F. Anti-MOG antibody: The history, clinical phenotype, and pathogenicity of a serum biomarker for demyelination. Autoimmun Rev 2016;15(4):307-24.
- Flanagan EP, Weinshenker BG. Neuromyelitis optica spectrum disorders. Curr Neurol Neurosci Rep 2014;14(9):483.
- Ohnari K, Okada K, Takahashi T, et al. Evoked potentials are useful for diagnosis of neuromyelitis optica spectrum disorder. J Neurol Sci 2016;364:97-101.
- Sellner J, Boggild M, Clanet M, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol 2010;17(8):1019-32.
- Bennett JL, de Seze J, Lana-Peixoto M, et al. Neuromyelitis optica and multiple sclerosis: Seeing differences through optical coherence tomography. Mult Scler 2015;21(6):678-88.
- Chen H, Qiu W, Zhang Q, et al. Comparisons of the efficacy and tolerability of mycophenolate mofetil and azathioprine as treatments for neuromyelitis optica and neuromyelitis optica spectrum disorder. Eur J Neurol 2017;24(1):219-26.
- Kira JI. Unexpected exacerbations following initiation of disease-modifying drugs in neuromyelitis optica spectrum disorder: Which factor is responsible, anti-aquaporin 4 antibodies, B cells, Th1 cells, Th2 cells, Th17 cells, or others? Mult Scler 2017:1352458517703803.