
Abstract
The ready access to brain imaging has resulted in an increased detection of incidentally discovered pituitary lesions. Radiological and post mortem studies report the prevalence of pituitary incidentalomas to be as high as 10%.1 Exclusion of secondary headache is a frequent clinical indication for brain imaging. It is therefore not uncommon to be faced with a patient with both headache and a pituitary abnormality. The clinician must decide if the pituitary lesion is of any relevance to headache or a purely incidental finding. The aim of this article is to review the association between pituitary tumours and headache, and to suggest a pragmatic approach to investigation and management.
Mechanism of headache in pituitary tumours
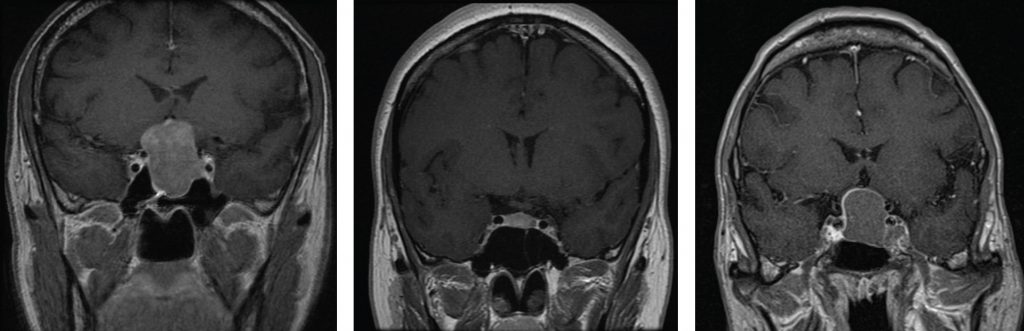
Pituitary tumours come to clinical attention as a result of their endocrine activity, the physical consequences of the lesion, or both. Whilst visual loss and hypopituitarism are clearly a result of compression of local structures, it is not clear if headache is a purely physical phenomenon. The traditional explanation of headache in pituitary tumours is dural stretch, but there is little evidence of an association between tumour size and headache.2 Large pituitary lesions can present with no headache at all (Figure 1), whilst small secretory micro-adenomas (<1 cm) may cause debilitating headache (Figure 2). Therefore, whilst headache is undoubtedly common in pituitary tumours, with a prevalence of 30-70%,3,4 the mechanism is far from clear.
The cavernous sinus contains the first and second branches of the trigeminal nerve and the internal carotid artery, which are potentially significant structures as regards headache (Figure 3). Despite this, prospective studies have shown no relationship between ipsilateral cavernous sinus invasion and headache.2,5 This further suggests that physical mechanisms are not a satisfactory explanation for pituitary headache. Despite these negative studies, the cavernous sinus cannot be completely dismissed as some pituitary tumour patients have cavernous sinus disease with severe ipsilateral headache.6 Headache with ipsilateral cavernous sinus invasion may have pronounced cranial autonomic features, which can dramatically improve after medical or surgical treatment.7-9 There are several reports of patients with macroprolactinomas invading the cavernous sinus presenting with ipsilateral refractory headache, which resolves within days of dopamine agonist treatment.7-9 Pituitary apoplexy is a specific situation whereby an acute vascular event within a pre-existing pituitary tumour gives rise to severe headache and diplopia. The mechanism of pain and cranial nerve palsy in apoplexy is probably via irritation of the 5th nerve, and 3rd, 4th and 6th nerves respectively.
There is no doubt that endocrine activity of the tumour can be relevant to pituitary headache. Acromegaly commonly has headache as an early feature, and this can be a useful clinical marker of disease activity. Somatostatin analogues, commonly used in the medical management of acromegaly, can have an immediate analgesic effect on headache. Interestingly, the control of headache and growth hormone (GH) suppression do not always go hand in hand, suggesting that the mechanism of somatostatin analgesia is not directly related to GH per se.10 It is hypothesised that pituitary headache may be caused by the secretion of an un-measured pro-nociceptive peptide that is suppressed by somatostatin. Prolactinoma and TSHoma patients may be associated with headache that is reproducibly aborted by somatostatin, supporting the idea of a hitherto unidentified pain-producing peptide being actively secreted in pituitary tumours.11
The hypothalamo-pituitary axis is known to be an important anatomical area in the pathophysiology of primary Trigeminal Autonomic Cephalgias (TACs). Functional imaging studies (fMRI and PET) demonstrate ipsilateral hypothalamic and cavernous sinus activation.12 Pituitary tumours have a higher prevalence of TACs than the general population,6 re-enforcing the view that this part of the brain is important in headache.
The genetic susceptibility of patients to headache may be as significant as tumour properties per se. A family history of headache is a predictive associative factor for pituitary headache.6 Therefore pituitary headache is likely to be a heterogeneous phenomenon dependent on the biochemical and physical characteristics of the tumour, as well as the genetic susceptibility of the patient to headache.
Pituitary incidentaloma
When an incidental pituitary lesion is discovered, it is important to rule out clinical signs of endocrine disease. Women should be asked about hyperprolactinaemic symptoms, including menstrual irregularity, fertility problems and galactorrhoea. Clinical signs of acromegaly and Cushing’s syndrome should be looked for. In early acromegaly subtle soft tissue signs may be present including carpal tunnel syndrome, increased snoring due to palatal oedema, and mild facial changes. Old photographs or previous self-images on mobile phones are useful to look for changes in appearance that patients and their families may not have recognised. In suspected Cushing’s syndrome, the presence of bruising and thinning of skin are particularly useful discriminatory features.
Serum prolactin is the most cost effective single test for a pituitary incidentaloma, a level > 1000 miU/L usually signifying a prolactinoma if no other causes of hyperprolactinaemia are found.13 In suspected acromegaly, a random GH and IGF-1 level is useful, although a formal OGTT with failure to suppress GH confirms the diagnosis. Assessment of thyroid status with fT4 and TSH will exclude TSH deficiency (low T4 with low or normal TSH) and TSHoma (high T4 and non-suppressed TSH). Screening tests for Cushing’s syndrome include 24h Urine Free Cortisol, overnight Dexamethasone Suppression Test (DST) or formal low dose DST, and are only needed if clinically indicated.
Clinical features of pituitary headache
The International Headache Society (I.H.S) Headache Classification System allows the clinician to formally classify headache attributed to hypothalamic or pituitary hyper- or hypo-secretion (I.H.S 7.4.4). It is useful to biologically phenotype the headache, because appropriate headache treatment will often lead to clinical response without the need to treat the pituitary lesion per se. The commonest headache phenotype in patients with pituitary tumours is chronic migraine.6 It is likely that the endocrine changes caused by a pituitary lesion trigger migraine in a predisposed individual. This is particularly common in young women with micro-prolactinoma, the same demographic as those predisposed to migraine. Hyperprolactinaemia commonly causes exacerbation of migraine via alteration in female hormones, rather than as result of any mass effect. The full range of TACs has been described in association with pituitary tumours, including cluster headache, SUNA, paroxysmal hemicrania and hemicrania continua, at a higher prevalence than the general population.4,6 TACs occur with small and large, non-functioning and functioning pituitary tumours and the precise mechanism is unclear. A sub-group of patients have headache that can only classified under I.H.S 7.4.4 and we have suggested modification of this to include the presence or absence of cavernous sinus invasion.6 Future studies are required to determine which specific clinical features are exclusive to pituitary tumours.
Management approach
The clinician must always consider that the pituitary lesion is incidental to headache. Standard pharmacological prophylactic or abortive headache treatment often leads to improvement in symptoms. If there are signs of endocrine excess, the pituitary lesion should be treated conventionally. Normalisation of endocrine status may lead to resolution of headache without the need for specific headache treatment. Dopamine agonist treatment of prolactinoma will usually lead to improvement of associated headaches. In acromegaly, surgical or medical treatment will often lead to abolition of headache, although somatostatin analogue over-use should be avoided.14 Surgical treatment of macro-adenoma will lead to improvement in headache in nearly 50% of patients.6 A difficult problem can be the patient with a pituitary macro-adenoma who presents with troublesome headache and ipsilateral cavernous sinus invasion. In this situation, headache should not be the sole indication for surgery, as there is no guarantee of resolution of symptoms. The usual indications for hypophysectomy are visual loss as well as endocrine control of the tumour. The American Endocrine Society lists unremitting headache as a relative indication for surgery,13 but it should be made clear to the patient that headache may not resolve post-operatively. Tumours that invade the cavernous sinus are relatively inaccessible surgically, even with the recent development of endoscopic surgery. Post-operative residual cavernous sinus disease should be managed in a multi-disciplinary setting both by the pituitary team and a dedicated pain or headache specialist. Potential therapeutic options include treatment of the tumour bulk itself with external beam or gamma knife radiotherapy, and specific management of the pain with the use of drugs or specific interventions to down-regulate trigemino-vascular pathway.
Summary
Pituitary tumours commonly present with headache and it is useful for the clinician to have a system for dealing with this problem. Full assessment of the headache phenotype as well as clinical and biochemical characterisation of the pituitary lesion are important to drive appropriate management. From an academic perspective, pituitary tumours may give interesting new insights into the pathophysiology of headache, and there is merit in studying this area more extensively.
References
- Scangas GA, Laws ER Jr. Pituitary incidentalomas. Pituitary. 2014;17(5):486-91.
- Levy MJ, Jäger HR, Powell M et al. Pituitary Volume and headache: size is not everything. Arch Neurol 2004;61:721-5.
- Dimpoulou C, Athanasoulia AP, Hanisch E, et al. The clinical characteristics of pain in patients with pituitary adenomas. Eur J Endocrinol 2014;171:581-91.
- Levy MJ. The Association of Pituitary Tumours and Headache. Curr Neurol Neurosci Rep 2011; 11:164-170.
- Abe T, Matsumoto K, Kuwazawa J et al. Headache associated with pituitary adenomas. Headache 1998;38:782-6.
- Levy MJ, Matharu MS, Meeran K et al. The clinical characteristics of headache in patients with pituitary tumours. Brain 2005;128(8):1921-30.
- Levy MJ, Robertson I, Howlett TA. Cluster headache secondary to macroprolactinoma with ispilateral cavernous sinus invasion. Case Rep Neurol Med 2012.
- Matharu MS, Levy MJ, Merry RT, Goadsby PJ. SUNCT syndrome secondary to prolactinoma. Neurol Neurosurg Psychiatry 2003;74(11):1590-2.
- Levy MJ, Matharu MS, Goadsby PJ. Prolactinomas, dopamine agonists and headache: two case reports. Eur J Neurol 2003;10(2):169-73.
- Levy MJ, Bejon P, Barakat M, Goadsby PJ, Meeran K. Acromegaly: a unique human headache model. Headache 2003;43(7):794-7.
- Williams G, Ball J,Lawson RA, Joplin GF, Bloom SR, Maskill MR. Analgesic effect of somatostatin analogue (octreotide) in headache associated with pituitary tumours. Br Med J (Clin Res Ed) 1987;295:247-8.
- May A, Bahra A, Buchel C, Frackowiak RS, Goadsby PJ. Hypothalamic activation in cluster headache attacks. Lancet 1998;352(9124):275-8.
- U.S Endocrine Society Clinical Practice Guideline 2011. Pituitary Incidentaloma.
- May A, Lederbogen S, Diener HC. Octreotide dependency and headache: a case report. Cephalalgia. 1994;14:303-4.