

An update on synucleinopathy pathogenesis
Recent and exciting developments are providing new clues as to how different synucleinopathies such as Parkinson’s Disease (PD), Multiple System Atrophy (MSA) and neocortical Lewy Body Dementia (LBD) might originate. Synucleinopathies comprise a large group of heterogeneous neurodegenerative disorders and although they present with distinct clinical phenotypes, they all share ɑ-synuclein (ɑ-SYN) protein deposits as a common histopathological hallmark. ɑ-SYN proteinaceous inclusions are found at specific predilection sites within the nervous system and their post-mortem confirmation is crucial for diagnosis of PD, DLB or MSA. In PD and DLB, ɑ-SYN inclusions are found in neuronal cell bodies and axons, which are termed Lewy Bodies (LB) and Lewy Neurites (LN), respectively. Patients that suffer from PD show predominant parkinsonism1 while patients with DLB exhibit progressive cognitive impairment as a predominant feature next to motor-related deficits or other core features.2,3 The clinical and histopathological features of MSA, however, are notably different compared to PD and DLB. In the central and peripheral nervous system of patients with MSA, ɑ-SYN does not only deposit in neurons but also in oligodendroglial and Schwann cells causing crescent-shaped glial cytoplasmic inclusions and filamentous Schwann cell cytoplasmic inclusions.4,5 Compared to PD and DLB, this translates to distinct symptomatology with corticospinal tract dysfunction, cerebellar ataxia and autonomic dysfunction that can exist next to a parkinsonian syndrome.6 The heterogeneity of synucleinopathies is also illustrated by the observation that these symptoms can overlap between patients, adding complexity to the diagnosis of these neurodegenerative diseases.
The reason why a single pathognomonic histopathological component is found in different diseases with different symptomatology has been a mystery for over several decades. In a link with prion diseases, researches hypothesised that differences in the ɑ-SYN aggregation process resulting in different ɑ-SYN aggregated structural isoforms might explain such a clinical heterogeneity. In transmissible encephalopathies or prion diseases such as bovine spongiform encephalopathy (BSE) or Creutzfeld Jakob’s Disease (CJD), different clinical phenotypes are observed because self-sustaining infectious protein particles, or prions, have the ability to encode structural information and replicate. These aggregated prion assemblies bear unique fingerprints and after exposure to the central nervous system (CNS) they incite specific biological and neurotoxic profiles. The pathogenic prion protein therefore bears intrinsic pathogenicity via a protein-based strain-encoded mechanism causing strain-specific symptomatology.
With this concept of prions in mind, researchers asked whether ɑ-SYN was also able to form strains and propagate in a similar manner. ɑ-SYN is a small and flexible protein of only a few kilodaltons that can readily aggregate in vitro from an unfolded, unstructured protein into megadalton β-sheet-rich fibrillar aggregates that consist out of several thousands of ɑ-SYN molecules (Figure 1). In an initial study performed by Bousset and colleagues it was shown that when ɑ-SYN assembles under different aggregation conditions, it is able to aggregate into distinct high-molecular weight fibrillar polymorphs.8 The resulting two ɑ-Syn fibrils were extensively characterised and appeared structurally different in various biophysical assays. They had the ability to encode structural information and faithfully propagate between cells, which suggested that these ɑ-SYN fibrils behaved as strains. The morphology of these two strains was examined by electron microscopy and it was shown that one strain appeared straight while the other was more twisted. This led the researchers to label these two types of strains as ‘fibrils’ and ‘ribbons’, respectively. Both fibrils and ribbons furthermore responded very different in biochemical and functional assays, indicating that the structural variations of the ɑ-SYN strains were responsible for the different biological phenotypes that they induced.
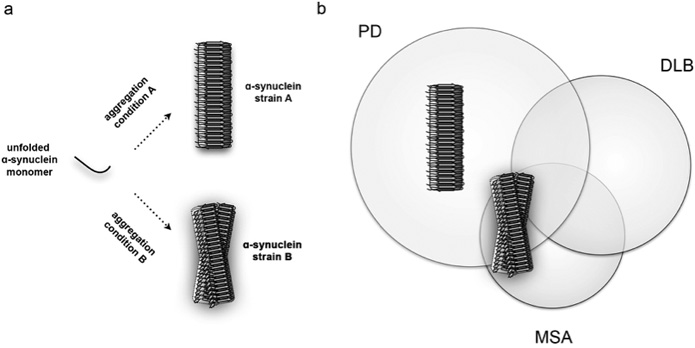
An important step in proving the relevance of these ɑ-SYN strains for synucleinopathy pathogenesis was to inoculate these strains in brain and study their potential pathogenicity in vivo. By using an experimental rat model that allowed to examine the inoculation of fibrils and ribbons in native and ɑ-SYN overexpressing backgrounds, it was shown that the two synthetic ɑ-SYN strains exhibited distinct histopathological, neurotoxic and behavioral phenotypes.9 Ribbons induced aberrant Lewy neurite-like inclusions in dopaminergic cells of the substantia nigra pars compacta, a severely affected midbrain nucleus in PD, while this was virtually absent for fibrils. In contrast, fibrils induced motor impairment in absence of clear dopaminergic cell death indicating that ɑ-SYN aggregates have to ability to impair synaptic activity and motor impairment before overt neurodegeneration takes place. When the two ɑ-SYN strains were inoculated in the brain of rats that overexpressed ɑ-SYN, neurotoxicity increased drastically for animals that were exposed to fibrils. This, however, was not the case for animals exposed to the other strain or even oligomers, which are smaller variants of aggregated ɑ-SYN that are assumed to play an important role in several neurodegenerative diseases. Another remarkable finding was that in ɑ-SYN overexpressing conditions, ribbons induced a second histopathological feature. Next to neuritic inclusions, ribbons promoted sparse ɑ-SYN inclusions in oligodendroglial cells, the histopathological hallmark of MSA. This was not observed for other conditions and reveals that strains can differentially promote different types of proteinaceous inclusions. These results therefore demonstrate that unique structural assemblies promote disease pathogenesis with different outcomes in histopathology, neurotoxicity and symptomatology.
By using synthetic strains, these experiments provided a proof-of concept as to how synucleinopathies might originate. To further examine whether the brain of patients with PD or MSA indeed contain different types of ɑ-SYN aggregates – the strains, the group of Stanley Prusiner assessed whether brain isolates derived from PD and MSA patients possessed ‘prion-like’ properties.10 They indirectly showed that brain isolates derived from patients with PD and MSA, which contain ɑ-SYN aggregates, might indeed be very different. In various cellular and in vivo assays, the isolated ɑ-SYN-enriched fractions were able to accelerate ɑ-SYN aggregation similar to what is observed for prions. These brain fractions furthermore induced strain-specific effects by lowering the incubation time during second passage experiments after inoculation of MSA-derived isolates in transgenic mice. These effects were only observed for post-mortem brain isolates derived from patients that were diagnosed with MSA but not with PD and indicates that the structural ensemble of ɑ-SYN within the brain of these two groups of patients might indeed be very different.
Prions can invade the CNS from the periphery and subsequently spread to anatomically connected regions.11 A horizontal transfer of prions can therefore occur between individuals, although only in extremely rare conditions, as result of blood transfusions, organ transplantation, injection of growth hormone extracts, exposure to improperly sterilised medical material or eating BSE-contaminated food.7 In parallel with this, several independent research groups assessed whether misfolded ɑ-SYN might also spread in a similar pattern for the periphery to the CNS. Strikingly, administration of ɑ-SYN aggregates into the blood, muscles or stomach of rodents successfully resulted in transmission of aggregated ɑ-SYN to the CNS.9,12,13 Synucleinopathy patients very often present with clinical symptoms that indicate a peripheral involvement. Lewy pathology is also present in the enteric nervous system of patients and a long prodromal stage precedes the typical symptoms that characterise PD, DLB or MSA. These findings collectively suggest that the spread of ɑ-SYN is not restricted to the CNS but extends far beyond.
A complete understanding of how ɑ-SYN misfolds and aggregates into different strains with central and peripheral involvement could lead to the development of new diagnostic tools and therapeutic strategies. PET tracers already exist to detect Aβ plaques in the brain of Alzheimer’s patients and solving the structure of ɑ-SYN strains might yield new opportunities to develop tracers for synucleinopathies as well. In addition, aggregated ɑ-SYN particles are present in the blood of PD patients.14 The exact properties of these aggregates, however, are unknown. If strains would propagate systemically, then immuno-based assays (e.g. ELISA) might yield interesting opportunities to detect different types of aggregates by analyzing cerebrospinal fluids or blood samples. The identification of disease-specific strains could furthermore allow differentiating between PD, DLB and MSA patients, who often show overlapping symptoms. Therapeutic strategies involve directly tackling ɑ-SYN aggregation via blocking ɑ-SYN aggregation, promoting the clearance of ɑ-SYN aggregates or immunization therapies. The existence of strains thus not only helps us to understand why synucleinopathies are so different, it also holds great promise in our continuous efforts to improve diagnostic tools and therapeutic strategies.
References
- Berg D et al. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov Disord 2014;29:454-62, doi:10.1002/mds.25844.
- McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. Journal of Alzheimer’s disease : JAD. 2006;9,:417-23.
- McKeith IG et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996;47:1113-24.
- Nakamura K et al. Filamentous aggregations of phosphorylated alpha-synuclein in Schwann cells (Schwann cell cytoplasmic inclusions) in multiple system atrophy. Acta Neuropathol Commun 2105;3:29, doi:10.1186/s40478-015-0208-0.
- Spillantini MG et al. Neurosci Lett 1998;251:205-8.
- Gilman S et al. Second consensus statement on the diagnosis of Filamentous alpha-synuclein inclusions link multiple syste m atrophy with Parkinson’s disease and dementia with Lewy bodies. multiple system atrophy. Neurology. 2008;71:670-6,doi:10.1212/01.wnl.0000324625.00404.15.
- Will RG. Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Brit Med Bull. 2003;66:255-65, doi:10.193/bmb/dg66.255.
- Bousset L et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575, doi:ncomms3575 [pii]10.1038/ncomms3575.
- Peelaerts W et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. 2015;522;340-4, doi:10.1038/nature14547.
- Prusiner SB et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci USA. 2015; E5308-5317, doi:10.1073/pnas.1514475112 (2015).
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552-61, doi:nrm2204 [pii]10.1038/nrm2204.
- Sacino AN et al. Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci USA. 2014;111:10732-7, doi:10.1073/pnas.
- Holmqvist S et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128:805-20, doi:10.1007/s00401-014-1343-6.
- El-Agnaf OM. et al. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J 2006;20:419-25, doi:20/3/419 [pii] 10.1096/fj.03-1449com.
- Nemani VM. et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. 2010;65:66-79, doi:10.1016/j.neuron.2009.12.023.
- Theillet FX. et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. 2016:doi:10.1038/nature16531.