


There have been major advances in the characterisation of frontotemporal dementia (FTD) over the past two decades culminating in the development of internationally accepted diagnostic criteria for each of the clinical subtypes. The behavioural variant of FTD (bvFTD) is characterised by changes in personality and behaviour which have now been clearly defined with levels of diagnostic certainty: possible, probable and definite (Table 1).
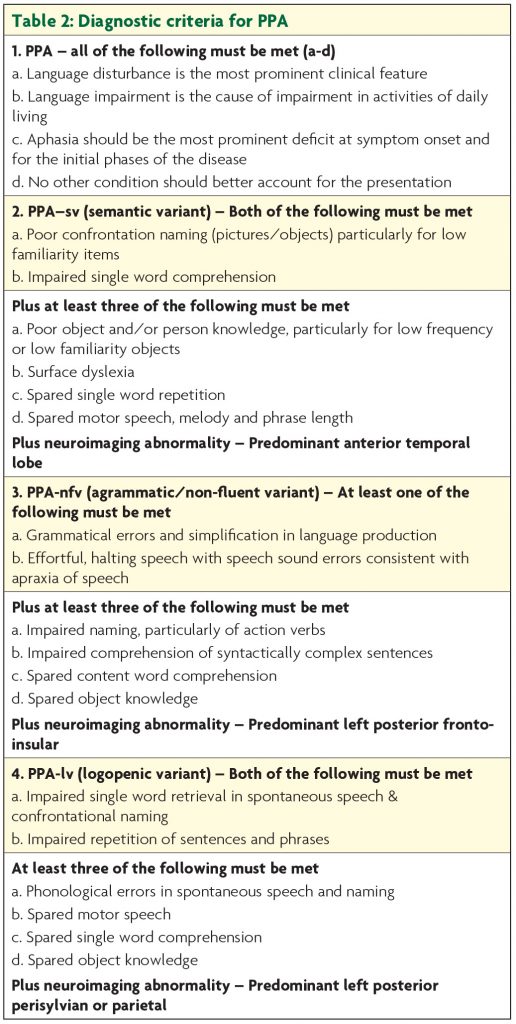
For cases presenting with progressive aphasia, three variants are now recognised: the semantic variant (semantic dementia) characterised by fluent speech with anomia and impaired single word knowledge in association with anterior temporal lobe atrophy; the non-fluent variant with apraxia of speech and/or agrammatism associated with inferior frontal atrophy; and the logopenic variant in which word finding difficulties and impaired verbal span predominate, and atrophy is centred around the angular gyrus (Table 2).
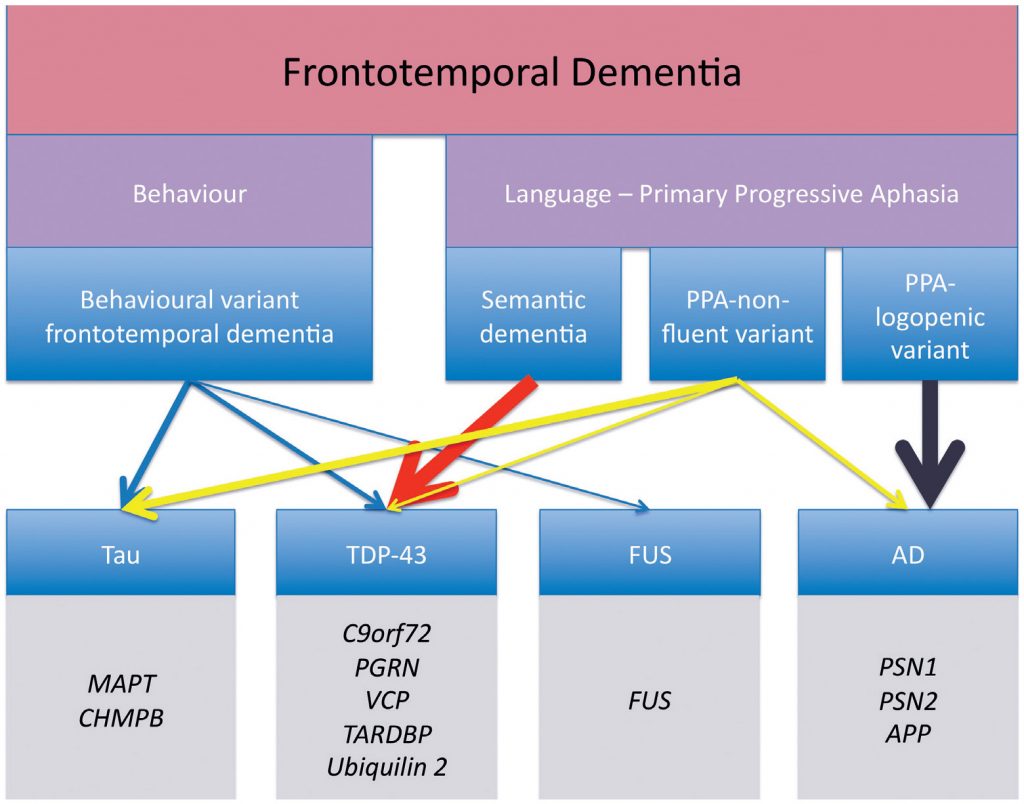
The distinction is not purely academic since underlying neuropathology is highly predictable. Those with semantic variant have TDP-43 type C and are rarely genetic. Those with the nonfluent form mostly have tau based FTD pathology, which may be genetic, and the logopenic form is associated predominantly with Alzheimer’s pathology,1,2 the pathology remains heterogeneous with an approximate 50-50 split between FTD-tau and FTD-TDP43 (Figure 1).
Developments in neuroimaging, notably functional MRI (fMRI), have heralded the concept of dysfunction within neural networks as the anatomical basis for abnormal behaviours and cognitive dysfunction in FTD. The salience network, comprising the insula, anterior cingulate cortex, amygdala, and a network of thalamic and subcortical structures, is involved early in the course of bvFTD and is implicated in the generation of social and emotional dysfunction.3 The link between FTD, deranged metabolism and abnormal eating behaviour is becoming clearer and seems likely to be linked to a complex neural network centred on the hypothalamus.4 Together these findings have encouraged FTD researchers to consider the contribution made by brain regions outside of the frontal and temporal cortices. The cerebellum, previously considered to be concerned primarily with motor function, has been implicated in a range of cognitive dysfunctions, and the thalamus, a key relay station for the signaling of sensory information and integration throughout the cortex, may also be involved.
Until recently MRI and Fluorine-18-Fluorodeoxyglucose-Positron Emission Tomography (FDG-PET) have been the imaging modalities of choice for the diagnostic work-up of patients with FTD. In more recent years, amyloid Pittsburgh compound B (PIB)-PET imaging has been useful to tease apart atypical cases when distinction from AD is clinically difficult. Following the success of amyloid PET imaging, researchers turned towards finding suitable tracers for tau protein and a number have been developed.5 Tau imaging has the potential to improve diagnostic accuracy in FTD through the identification of tauopathies during life, whereas currently the underlying pathology can be identified post-mortem only. This may allow more accurate case selection for clinical trials and subsequent pharmacological therapies. Tau PET imaging could also improve disease staging, as tau burden is closely linked with cognitive impairment, and determine the role of tau deposition in the preclinical stages of neurodegeneration. Although the potential benefits of tau imaging for clinicians, researchers and patients are clear there are issues to be resolved before the ideal ligand is identified and introduced to clinical practice.
It seems likely that future research will focus on in-vivo identification of other pathological proteins notably trans-activating responsive (Tar) sequence DNA binding protein (TDP-43). The discovery of TDP-43 consolidated the overlap between FTD and MND given that this protein is found in a proportion of those with FTD and the vast majority of familial and sporadic MND cases.6 Clinical overlap between these conditions has long been recognised but only fairly recently has the concept of an FTD-MND disease continuum become widely accepted.7
Cognitive/behavioural deficits may develop in parallel with motor deficits although either can occur initially. Cognitive and behavioural abnormalities are reported to occur in 50-75% of MND cases while approximately 15-25% of patients meet criteria for FTD.7 Of all the cognitive functions, executive function has received the most attention in MND that may have consequences for financial, medical and end of life decisions. Language deficits may be as common as executive dysfunction and adds another level of complexity to communication issues for MND patients. Similarly, social and emotional cognition domains are affected and behaviour is impaired, specifically a degree of apathy is found in up to 80% of patients, and disinhibition, lack of empathy and rigidity are also present.8 Conversely around 10% of FTD cases develop frank MND but subclinical motor features can be found in a much higher proportion of cases. Patients with the FTD-MND overlap syndrome tend to have the shortest survival of all FTD syndromes with death occurring within two to three years.
In 2011 the concept of the FTD-MND continuum was cemented, with identification of the C9orf72 genetic expansion on chromosome 9p21.1.9 This genetic expansion, a hexanucleotide GGGGCC repeat found on the non-coding region of chromosome 9, is pathogenic at greater than 30 repeats with most patients having repeat lengths in the thousands. Studies of affected carriers, asymptomatic carriers and their family members will provide further insight into this gene defect which, may have features in common with other repeat disorders. The exact penetrance is still unknown but it is believed that it is not fully penetrant, as unaffected elderly carriers have been identified. Three pathological mechanisms have been proposed to cause disease in C9orf72 carriers: loss of function of the protein encoded by the gene, toxic effects of RNA products which aggregate in the cell and toxicity caused by dipeptide repeat proteins. The C9orf72 expansion has been identified with mutations in other well-known causative genes in FTD including GRN and MAPT,10,11 which has led researchers to hypothesise that these genes and others may play a modifying role in C9orf72 expression. This expansion accounts for approximately one-third of familial FTD and up to 75% of familial FTD-MND cases. Notably, a significant minority (5-20%) of patients with apparently sporadic bvFTD also have the expansion.12 Collectively the three major genes, C9orf72, GRN and MAPT account for over 50% of familial FTD cases indicating there are clearly gene mutations yet to be discovered.
There is marked geographical variation in prevalence of the C9orf72 expansion with high rates of the expansion found in northern European countries, while remaining rare in Asian populations. Across the clinical spectrum of FTD, the predominant phenotype associated with the C9orf72 expansion is bvFTD, often occurring with features of MND, although non-fluent variant PPA cases have been reported. C9orf72 positive patients can be distinguished from negative cases based on a family history of MND, Parkinsonism and prominent psychosis at presentation.12 Delusions and hallucinations are generally rare in FTD but are a frequent presenting feature of the C9orf72 expansion some of who have a long history of psychiatric illness. A distinctive neuroanatomical signature has also emerged, with generally mild atrophy involving the thalamus and cerebellum in addition to the typical orbitomedial frontal and anterior temporal atrophy seen in bvFTD.13 A proportion of patients labelled as ‘slow-progressors’ or ‘phenocopy’ cases habour the C9orf72 expansion and in a study from our centre the proportion of possible bvFTD cases with the mutation was higher than found in cases with probable bvFTD.14
With regard to therapies for FTD, disease-modifying treatments have so far focused on tau pathology. Recent attempts to inhibit tau phosphorylation using lithium and tideglusib have failed, however a methylene blue derivative, leucomethylthioninium, shows promise as an inhibitor of tau aggregation and phase III clinical trials are currently underway. The discovery of the C9orf72 expansion has led to the theory that antisense oligonucleotide therapy, which show activity against toxic RNA effects as seen in the C9orf72 expansion, may be effective in the treatment of MND-FTD.15
In summary, the last few decades have seen rapid advances in our understanding of FTD encompassing clinical, neuroimaging, genetic and pathological fields with hopefully even more exciting discoveries on the horizon. Better awareness of FTD, together with the development of diagnostic criteria, has facilitated earlier diagnosis to ensure that these patients have timely access to necessary care and support although FTD is still poorly recognised in non-specialist settings. The identification of the C9orf72 expansion has challenged the concept of MND and FTD as single disease entities by explaining the genetic link between the conditions, and provides a platform to study the complex underlying molecular pathogenesis of these diseases.
References
- Mesulam M, Wicklund A, Johnson N, Rogalski E, Léger GC, Rademaker A, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Annals of neurology. 2008;63(6):709-19.
- Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006-14.
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62(1):42-52.
- Ahmed RM LS, Bartley L, Irish M, Kiernan MC, Hodges JR, Piguet O. Eating behavior in frontotemporal dementia: Peripheral hormones vs hypothalamic pathology. Neurology. In press.
- Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC. Tau imaging: early progress and future directions. The Lancet Neurology. 2015;14(1):114-24.
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science. 2006;314(5796):130-3.
- Lillo P, Savage S, Mioshi E, Kiernan MC, Hodges JR. Amyotrophic lateral sclerosis and frontotemporal dementia: A behavioural and cognitive continuum. Amyotrophic lateral sclerosis : official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2012;13(1):102-9.
- Lillo P, Garcin B, Hornberger M, Bak TH, Hodges JR. Neurobehavioral features in frontotemporal dementia with amyotrophic lateral sclerosis. Archives of neurology. 2010;67(7):826-30.
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A Hexanucleotide Repeat Expansion in< i> C9ORF72</i> Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron. 2011;72(2):257-68.
- Lashley T, Rohrer JD, Mahoney C, Gordon E, Beck J, Mead S, et al. A pathogenic progranulin mutation and C9orf72 repeat expansion in a family with frontotemporal dementia. Neuropathology and applied neurobiology. 2013.
- van Blitterswijk M, Baker MC, DeJesus-Hernandez M, Ghidoni R, Benussi L, Finger E, et al. C9ORF72 repeat expansions in cases with previously identified pathogenic mutations. Neurology. 2013;81(15):1332-41.
- Devenney E, Hornberger M, Irish M, Mioshi E, Burrell J, Tan R, et al. Frontotemporal dementia associated with the C9ORF72 mutation: a unique clinical profile. JAMA neurology. 2014;71(3):331-9.
- Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain : a journal of neurology. 2012;135(Pt 3):736-50.
- Devenney E, Hoon C, O’Callaghan C, Kumfor F, Hornberger M, Kwok JB, Halliday GM, Kiernan MC, Piguet O, Hodges JR. Progression in behavioural variant frontotemporal dementia: a longitudinal study. JAMA neurology. 2015:1-9
- Riboldi G, Zanetta C, Ranieri M, Nizzardo M, Simone C, Magri F, et al. Antisense oligonucleotide therapy for the treatment of C9ORF72 ALS/FTD diseases. Molecular neurobiology. 2014;50(3):721-32.