
Abstract
Narcolepsy is thought to affect 0.05% of Caucasian populations and frequently causes severe symptoms across the 24-hour period. It is best viewed as a disorder of sleep-wake regulation with particular abnormalities of the rapid eye movement (REM). Typical cases are due to specific loss of a subset of hypothalamic neurons containing the neuropeptide, hypocretin (orexin). Several lines of evidence, including a causal link to the swine ‘flu vaccination, suggest autoimmune destruction of these neurons as an initial event.
Narcolepsy is now classed as either type 1 or type 2, depending on sleep investigation results and whether cataplexy and/or hypocretin deficiency is present. However, it is likely the classification system will be further refined.
Treatment options remain symptomatic and are often only partially effective. A new wake-promoting agent that increases brain histamine levels (pitolisant) has recently become available and will probably be used alongside modafinil and more traditional psychostimulants such as dexamphetamine. Powerful hypnotic agents, notably, sodium oxybate, consolidate the fragmented sleep frequently seen in narcolepsy and improve many of the daytime symptoms as a likely consequence.
Key points
- The narcoleptic syndrome is best viewed as a disorder of sleep-wake regulation, particularly affecting the REM sleep stage. Its phenotype is wide and includes elements not directly related to sleep such as appetite control, perhaps reflecting hypothalamic dysfunction.
- Nocturnal sleep fragmentation is a key feature and helps to explain why the sedative agent, sodium oxybate, is the best available treatment.
- The most recent diagnostic classification divides narcolepsy into type 1 (with cataplexy and significant hypocretin deficiency) and type 2 (without cataplexy and normal or low hypocretin levels). In these latest guidelines, the multiple sleep latency test remains an important diagnostic tool despite its poor sensitivity and reliability.
- A recent surge in incidence amongst children in particular following the swine ‘flu vaccination (Pandemrix) in 2009 has fuelled the notion of an autoimmune aetiology although many questions remain.
- Future treatments are likely to focus on hypocretin replacement via oral or intra-nasal medication. The newest useful treatment to become available is a novel stimulant drug that increases cortical histaminergic transmission, Pitolisant.
Introduction
Despite major advances in our understanding of narcolepsy and its neurobiology over the last 15 years, many questions concerning its nature and causation remain. Furthermore, the remarkable landmark discovery that specific loss of around 70000 neurons in the lateral hypothalamus containing the neuropeptide hypocretin could cause human narcolepsy and cataplexy1 has yet to lead to any significant therapeutic breakthroughs. Nevertheless, study of the hypocretin system in the brain has provided significant insight into how the sleep-wake cycle is regulated as well as furthering the diagnostic process in narcolepsy.
Given that narcolepsy reflects a neurochemical deficiency with a presumed spectrum of severity, perhaps it is not surprising that narcoleptic symptoms also vary between patients. In general, however, the adverse effects of narcolepsy on quality of life are increasingly recognised and most patients have equivalent measures of disability to those with treatment-resistant epilepsy.2 Moreover, narcolepsy typically affects young subjects, is life-long and also associated with numerous co-morbidities.
Promising research in several animal models of narcolepsy suggests that pharmacological replacement of hypocretin as a specific and effective pharmacological treatment remains a viable goal.
Defining the narcoleptic syndrome
Narcolepsy is now best viewed as a syndrome of severe sleep-wake dysregulation, particularly with respect to rapid eye movement (REM) sleep (Figure 1).
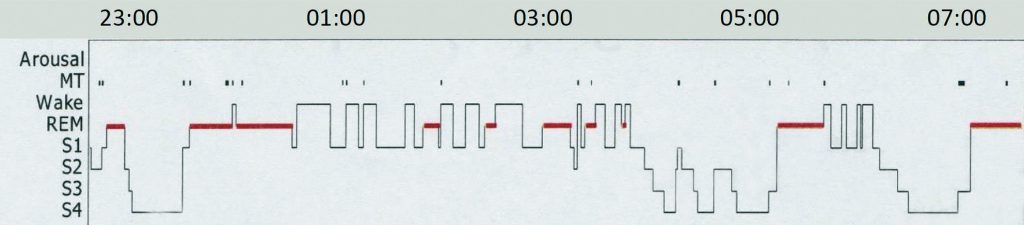
Although small in number, excitatory hypocretin-containing neurons project to numerous key brain areas crucial for behavioural state control. Without these neurons, the brain is far less able to maintain or consolidate either full wakefulness or, indeed, the state of sleep. Subjects may spend significant portions of the day somewhere in the spectrum between wake and sleep with reduced alertness or concentration as major features. Study of narcoleptic patients has furthered the concept of “localised” sleep, occurring independently in discreet parts of the brain. Indeed, elements of normal REM sleep intruding into the predominantly wakeful state, such as bizarre visual imagery or voluntary muscle paralysis, are key clinical diagnostic features for narcolepsy.
Cataplexy remains by far the most specific symptom in narcolepsy and affects around 70% of subjects. The wide spectrum of symptom severity is increasingly acknowledged with some patients reporting simply an inability to deliver punchlines of jokes with apparent speech arrest. Precisely why emotions or their anticipation should trigger neural activity in descending (glycinergic) pathways inhibitory to motor neurons, a feature of normal REM sleep, remains a fascinating conundrum. Cataplexy in children is now recognised as often having a distinct phenotype to the adult form. In particular, localised facial weakness is more apparent, often accompanied by grimacing, tongue protrusion or other “positive” motor phenomena which may lead to diagnostic confusion.3
Prior to changes in the latest diagnostic guidelines (International Classification of Sleep Disorders, ICSD-3),4 it was possible to diagnose narcolepsy on clinical grounds alone if typical cataplexy was present in the presence of persisting daytime somnolence. However, investigations are now required for formal diagnosis and distinction is made between type 1 and type 2 narcolepsy (see Table 1).
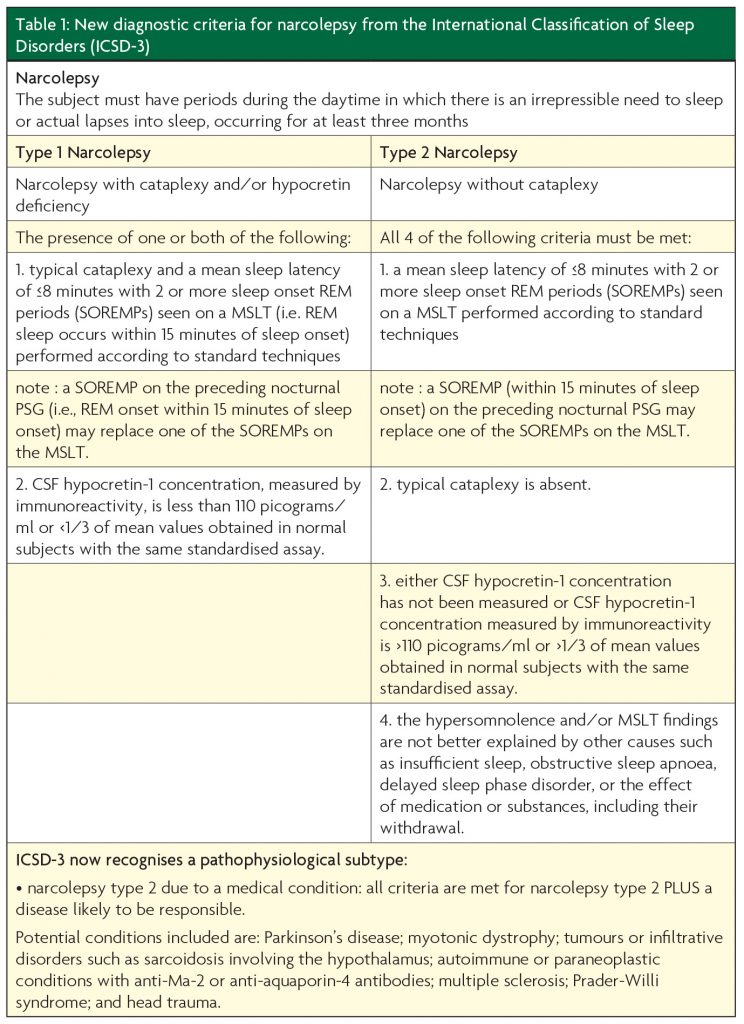
The emphasis on the multiple sleep latency test (MSLT), especially in type 2 narcolepsy, has led to major concerns, given its poor diagnostic sensitivity and specificity. Furthermore, it is a test very prone to protocol violations and associated difficulties with interpretation such that many clinicians would argue narcolepsy without cataplexy, in particular, should remain predominantly a clinical diagnosis, perhaps supported by investigations. Some evidence is emerging that partial hypocretin deficiency may explain many cases of type 2 narcolepsy.5
A number of symptoms and medical issues not obviously or directly related to sleep are now recognised in narcolepsy. Many patients have dysregulation of appetite control and admit to severe food cravings, usually at night particularly for sweet flavoured items. Not infrequently, nocturnal eating occurs without conscious control or full awareness as an apparent non-REM sleep parasomnia that may accompany narcolepsy. As a possible consequence of disordered appetite control, rather than reflecting relative physical inactivity, obesity is significantly commoner in narcoleptic populations even though evidence suggests they eat less per day than control populations.6 Whether this reflects a metabolic disorder, perhaps related to abnormal control of hypothalamic satiety hormones such as leptin, remains to be established. Similarly, although poorly studied, narcoleptic subjects also often report marked post-prandial sleepiness, particularly after large unrefined carbohydrate meals. Manipulations of diet can therefore sometimes help improve general alertness.
Male narcolepsy patients, in particular, who put on significant weight in middle-age are at significant risk of obstructive sleep apnoea syndrome (OSAS) which further complicates their sleep-wake control. Deteriorating control of daytime sleepiness in an obese subject, previously well controlled with daytime stimulant therapy, might suggest this additional sleep disorder. Unfortunately, even if correctly diagnosed with OSAS, narcolepsy patients tend to tolerate ventilation masks very poorly, often due to dream-like or hallucinatory intrusions involving the mask itself. If so, additional nocturnal sedation may allow better compliance.
It seems very likely that mood disorders are much commoner in narcolepsy patients both as a likely reaction to the disruptive effects of the syndrome and the accompanying features of sleep deprivation. The input of psychiatric expertise can therefore be useful although, in the author’s experience, care must be taken not to interpret REM sleep-related phenomena, particularly hypnagogic hallucinations without a delusional component, as primary psychotic features.
Generalised pain syndromes resembling fibromyalgia are often a prominent concern in narcolepsy patients. The bi-directional relationship between sleep disruption and pain perception may largely explain this observation.7 Neuropathic pain agents such as gabapentin are generally more useful than routine analgesics and much preferred to opiates which invariably disrupt the sleep-wake cycle and control of nocturnal breathing. Restless legs syndrome also appears particularly severe in some patients and may merit specific therapy with low dose dopaminergic agonists, especially if associated periodic limb movements are prominent overnight.
Theories of causation
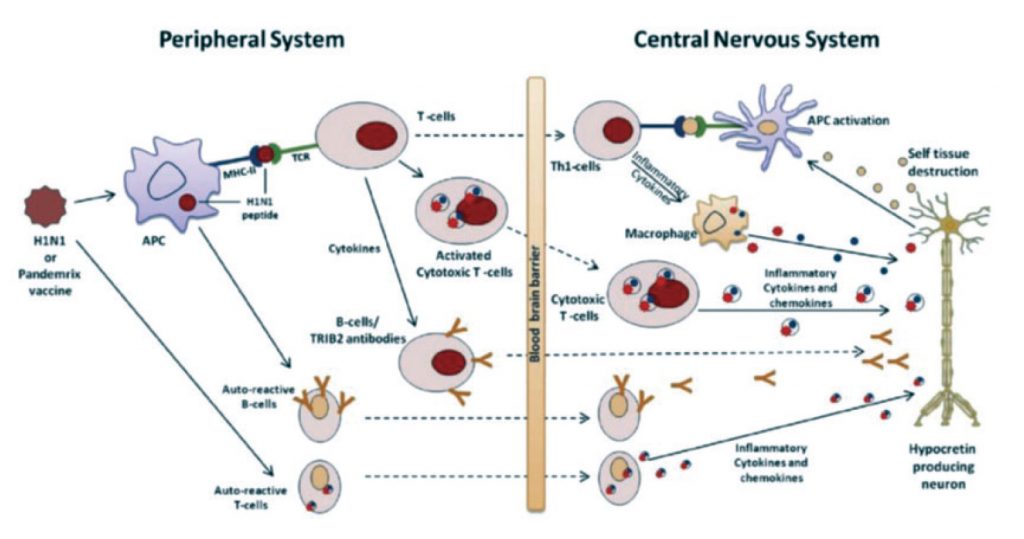
The fact that typical type 1 narcolepsy has one of the tightest HLA associations of any disease has fostered theories of an autoimmune aetiology for some time even though there are no clear links to other autoimmune conditions. Over recent years, however, a number of associations with presumed pathogenic antibodies have been proposed without clear subsequent substantiation.8 A reliable relationship to T cell receptor polymorphisms in narcoleptic patients has suggested that cell mediated destruction of hypocretin neurons may be an important mechanism behind cell death.9 Disappointingly, attempts to treat narcolepsy with various immunomodulatory agents have generally been unsuccessful in the absence of controlled trials. A recent case report suggesting an impressive clinical response to a monoclonal antibody, prescribed for an incidental lymphoma, is intriguing, however.10
Evidence from several countries that the swine ‘flu vaccine, Pandemrix, given to several million people in 2009 and early 2010 led to an abnormal surge in childhood and, to a lesser degree, adult cases of typical narcolepsy has further fuelled the “autoimmune” theory.11 If correct, however, it remains unclear whether the pathogenic process reflects a reaction to the strong adjuvant chemicals added to the vaccine or is simply molecular mimicry, related to proteins within the vaccine or indeed the virus itself. Equally unexplained is the frequent considerable delay between any proposed vaccine-related inflammatory reaction in the hypothalamus and narcolepsy symptom onset. One speculative theory is that any initial minor damage to the hypocretin neurons may promote a subsequent slow degenerative process, perhaps by an excitotoxic mechanism given the extremely high metabolic energy demands of these particular neurons.
Further insight into the potential vulnerability of hypocretin neurons is likely to come from a rare autosomal dominant genetic disorder reported in several families with a DNMT mutation.12 Although the phenotype is a little variable, particularly regarding symptom severity, many affected individuals have typical narcolepsy type 1 with cataplexy and associated hypocretin deficiency as part of their clinical picture. Other features may include myoclonus, deafness, ataxia and cognitive decline, superficially resembling a mitochondrial disorder and perhaps supporting an explanation of specific neuronal damage in neurons with particularly high energy demands
New and future treatments
Delays both in the diagnosis of narcolepsy, often years after symptom onset, and any significant loss of hypocretin neurons after an initial putative immune-related insult mean that standard immunosuppressive therapy is unlikely to be effective or practicable. The future realistic goal for more effective treatment probably lies with hypocretin replacement, ideally via an oral agent. To date, rodent and canine models of narcolepsy due to hypocretin deficiency or receptor mutations have demonstrated good responses to a variety of techniques that increase brain levels of hypocretin, fuelling hope for the human condition.13 Unfortunately, hypocretin’s neuropeptide structure makes it difficult to develop oral agonists that will penetrate into the brain although effective oral antagonists have been produced as treatments for insomnia. Intra-nasal hypocretin has produced promising data in sleep-deprived primates but the clinical data relating to narcolepsy has been disappointing so far.
The conventional approach to narcolepsy treatment is to prescribe daytime psycho-stimulants that are thought to activate the catecholaminergic component of the ascending reticular activating system. These drugs may suppress cataplexy in addition to improving daytime alertness although agents to suppress REM sleep, typically anti-depressant drugs such as venlafaxine, may be needed to treat the former. Modafinil with or without supplemental amphetamine or amphetamine-like drugs, such as methylphenidate, would be a typical wake-promoting combination. These agents are thought to have a dopaminergic action, either directly or indirectly, although Modafinil’s precise mechanism remains obscure, despite having been used as a first-line therapy for 17 years. A newly developed psychostimulant with a novel mode of action on central histaminergic systems is likely to become a useful additional agent to improve daytime alertness.15 Around 70000 histaminergic neurons in the anterior hypothalamus exert a powerful cortical excitatory effect that promotes wakefulness via H1 receptors. Histamine activity correlates well with the active or wakeful state and is generally lower than normal in narcolepsy. Pitolisant is a selective histamine antagonist which inhibits H3 autoreceptors in the hypothalamus, effectively promoting cortical histamine release. The drug has recently gained approval and early experience suggests that it will be a useful addition to currently available stimulant therapy.
Over the last decade, increasing attention to improving overnight sleep in narcolepsy has produced significant therapeutic advances. In particular, the controversial drug, sodium oxybate, usually given in divided doses overnight has been shown by trial data and in clinical practice to be the most effective single drug available.14 Aside from consolidating nocturnal sleep and enhancing its deeper stages, sodium oxybate often abolishes cataplexy within a few months of use as well as significantly improving daytime somnolence. Given its high price, issues over cost effectiveness have unfortunately severely limited its availability as have concerns over its potential misuse in society, primarily as a “date rape” drug.
References
- Dauvilliers Y, Arnulf I, Mignot E. Narcolepsy with cataplexy. Lancet 2007; 369:499-511.
- Broughton WA, Broughton RJ. Psychosocial impact of narcolepsy. Sleep 1994; 8(suppl):S45-9.
- Rocca FL, Pizza F, Ricci E, Plazzi G. Narcolepsy during childhood: an update. Neuropaediatrics 2015;46:181-98.
- Sateia MJ. International classification of sleep disorders – third edition: highlights and modifications. Chest 2015;146:1387-94.
- Andlauer O, Moore H 4th, Hong SC et al. Predictors of hypocretin (orexin) deficiency in narcolepsy without cataplexy. Sleep 2012;35:1247-55.
- Heier MS, Jansson TS, Gautvik KM. Cerebrospinal fluid hypocretin 1 deficiency, overweight, and metabolic dysregulation in patients with narcolepsy. J Clin Sleep Med 2011;7:653-8.
- Roehrs T, Roth T. Sleep and pain: interaction of two vital functions. Semin Neurol 2005;25:106-16.
- Lim AS, Scammell TE. The trouble with Tribbles: do antibodies against TRIB2 cause narcolepsy? Sleep 2010;33:857-8.
- Hallmayer j, Faraco J, Lin L et al. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat Genet 2009;41:708-11.
- Donjacour CE, Lammers GJ. A remarkable effect of alemtuzimab in a patient suffering from narcolepsy with cataplexy. J Sleep Res 2012;21:479-80.
- Partinen M, Kornum BR, Plazzi G et al. Narcolepsy as an autoimmune disease: the role of H1N1 infection and vaccination. Lancet Neurol 2014;13;600-13.
- Baets J, Duan X, Wu Y et al. Defects of mutant DNMT1 are linked to a spectrum of neurological disorders. Brain 2015;138:845-61.
- Slack SW, Yamanaka A, Kilduff TS. Challenges in the development of therapeutics for narcolepsy. Prog Neurobiol 2015; S0301-0082(15)30023-X. doi :10.1016/jpneurobio.2015.12.002 [epub ahead of print]
- Alshaikh MK, Tricco AC, Tashkandi M et al. Sodium oxybate for narcolepsy with cataplexy: systematic review and meta-analysis. J Clin Sleep Med 2012;8:451-8.
- Dauvilliers Y, Bassetti C, Lammers GJ et al. Pitolisant versus placebo or modafinil in patients with narcolepsy: a double-blind, randomised trial. Lancet Neurol 2013;12:1068-75.