



Introduction
Parkinson’s disease (PD) is a heterogeneous, neurodegenerative disorder affecting 6.3 million people worldwide and 1.2 million in Europe.1 The annual cost to Europe is estimated at 13.9 billion euro2,3 and the numbers are predicted to double by 2030.4 PD places a huge socioeconomic burden on Western economies and poses a major challenge for patients and society. Only symptomatic treatment is available. Traditional linkage analysis, gene cloning and Genome Wide Association Studies (GWAS) have identified several loci and genes associated with monogenic PD. The study of monogenic forms of PD may lead to the identification of new targets for pharmacotherapy and these may ultimately translate into new therapies for sporadic PD. However, treatment developed using these new technologies may only be effective for specific genetic mutations eg. kinase inhibitors for LRRK2 mutations. GWAS and linkage analysis have identified 18 Parkinson’s disease loci (PARK) numbered in a chronological order. This classification is imperfect as it contains both confirmed and unconfirmed loci (loci not replicated) and the causative gene remains unknown for many loci. Additionally one of the proposed loci was later found to be previously reported (PARK4 and PARK1).5
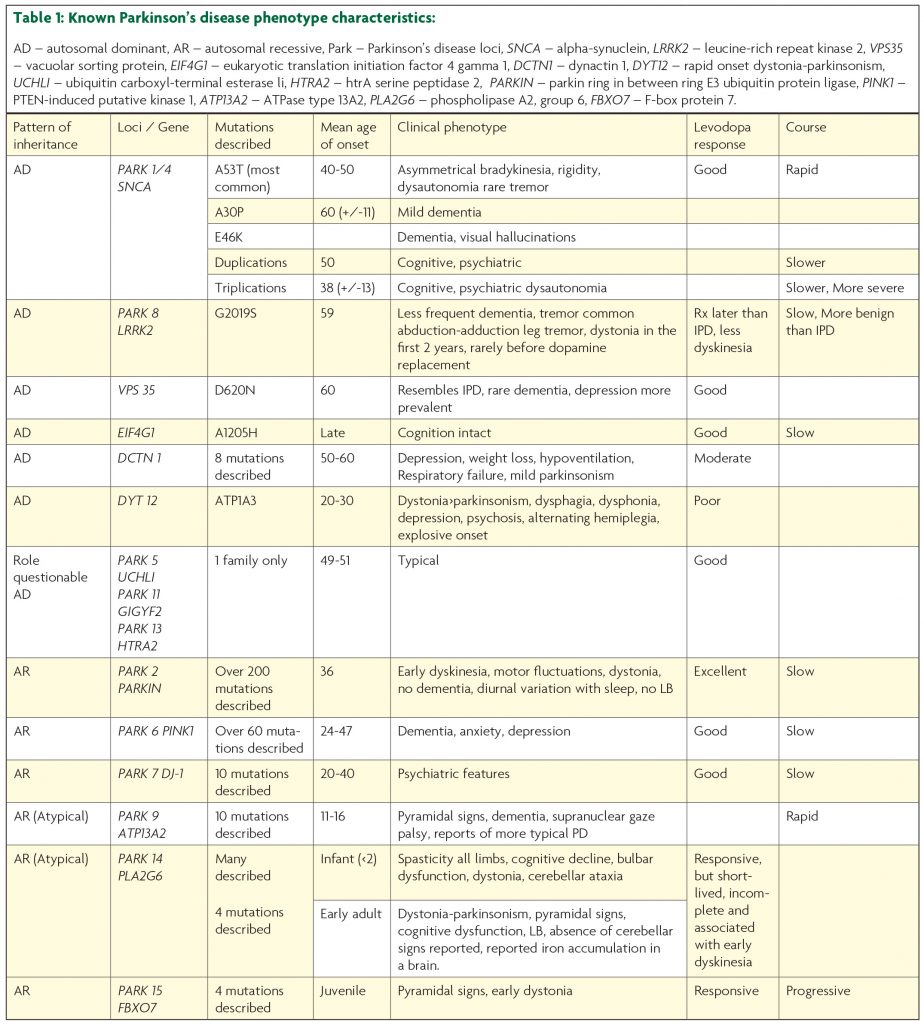
Mutations in seven genes cause either autosomal dominant (SNCA, LRRK2, VPS35), or autosomal recessive (Parkin, DJ1, PINK1, ATP13A2) familial PD.5 Moreover some of these genes contain polymorphisms which act as risk factors for the development of PD.6
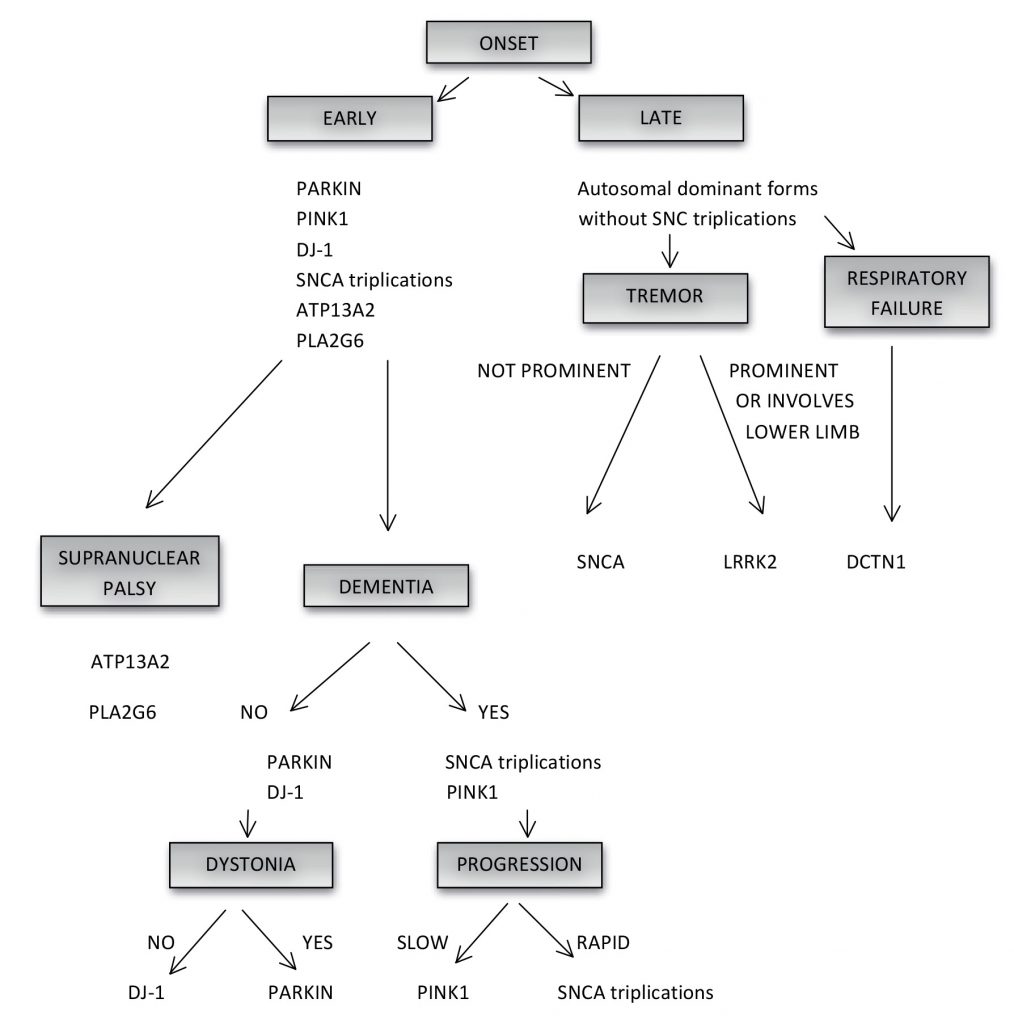
Clinical signs can be used to suggest ‘typical’ or ‘atypical’ forms of PD. Age-of-onset is used to classify PD into juvenile-onset (< 20 years), early-onset (between 20 and 40 years) and late-onset (>60 years) disease (Figure 1). The majority of patients have sporadic disease.6 Although the true Mendelian forms of PD are rare (occuring in 30% of familial and 3-5% of sporadic PD)5 there is a positive family history in 10% of patients with apparently sporadic PD.6
The pattern of inheritance may be elusive at times e.g. reduced penetrance associated with autosomal dominant inheritance may mimic recessive disease. In addition, heterozygote mutations in certain ‘recessive’ genes have been associated with late onset disease, possibly because of partial expression of the corresponding protein.5
Autosomal dominant mode of inheritance: (Table 1)
- SNCA (PARK1/PARK4), OMIM: 163890
In 1997, the first mutation linked to autosomal dominant monogenic PD was reported by Polymeropulous.7 The mutation (A53T) in a large Italian kindred (the Contoursi kindred) occurred in the synuclein gene (SNCA). SNCA encodes α (alpha)-synuclein (PARK 1/PARK 4) which is a small, 140-amino-acid protein8 abundant in Lewy bodies. Subsequently two other point mutations A30P (described in one German family) and E46K (one Basque family) were described.5,9 All three mutations are very rare, with A53T being the most common (reported in one Italian, eight Greek, two Korean and one Swedish families).5,9 Aside from point mutations, multiplications of SNCA may also occur. Duplications were found in thirteen families with monogenic PD and four sporadic PD cases, while triplications were described in three independent families.5
The age-of-onset in the A53T families is typically between 40 and 50 years of age, and is associated with asymmetrical bradykinesia, rigidity,7 good levodopa-responsiveness, dysautonomia, and less commonly rest tremor.10 The course of the disease is rapidly progressive (the interval from the onset to death is 10 years) (Table 1).
The A30P mutation is associated with a later age-of-onset (60 years (+/-11)), and mild dementia may occur.11 The E46K mutation is linked with dementia and visual hallucinations (dementia with Lewy bodies)11 (Table 1).
Disease-onset associated with duplications occurs at around 50 years and the age-of-onset associated with triplications is earlier at 38 years (+/-13).10 Therefore a gene dosage effect is present, in which triplications are associated with a 100% increase in protein expression resulting in a younger age-of-onset and more rapid disease progression. In comparison duplications are associated with a 50% increase in protein expression12 and later-onset monogenic PD with slower disease progression and cognitive and psychiatric decline.10 Triplications are commonly linked with dysautonomia.13 Therefore SNCA duplications and triplications should be considered in patients with an early-onset Lewy body disease picture in which early cognitive impairment and autonomic dysfunction is seen (Figure 1).
In 2013 two new SNCA point mutations were reported: H50Q and G51D. The H50Q mutation was described in a British patient with onset at 71 years and a Canadian-British patient origin with onset at 60 years. Disease was characterised by good levodopa-response and cognitive decline.11,14 The G51D mutation was reported in seven patients from British and French families and it was associated with dysautonomia, pyramidal signs, depression, anxiety,10,11,14 and cognitive decline.12 Age-of-onset was before 40 years, levodopa-response was moderate, and the monogenic PD had a rapid course with death within 10 years.12
- LRRK2 (Leucine-rich repeat kinase 2) (PARK8), OMIM: 609007
LRRK2 mutations in PD were identified in 2004 and subsequently were found to be the most common cause of autosomal dominant monogenic PD. LRRK2 is a large gene with 54 exons, encoding a 2527 amino-acid protein.13 It is responsible for 5-15% of autosomal dominant familial, monogenic PD and approximately 1% of sporadic PD in Caucasian patients.6 Previous studies in Ireland reported 1 mutation in 236 sporadic patients (<1%) and 1 mutation in 35 familial patients (3%).15 However, our follow up study of 133 Irish patients with familial PD found only 2 patients (1.5%) with LRRK2 mutations and 1 (0.26%) patient of 388 in sporadic PD.16,17 Therefore we found 3 (0.58%) patients with LRRK2 mutation in 521 patients with familial and sporadic PD.
Over 100 gene variants18 have been described with 7 mutations proven to be pathological.12 The most common mutation is G2019S12 and is most prevalent in the Middle East, Portugal, Spain and Italy with a north-south gradient evident in Europe.12,15,18 In Ashkenazi Jews the frequency is 29.7% in familial cases and 13.3% in sporadic cases and increases to approximately 40% in sporadic and in familial cases in North African Berbers.6,15 Onset is by age 59 with slower progression, less frequent dementia, and a more benign course than sporadic PD6 (Table 1). Tremor is the cardinal symptom, reported in 60-99% of patients.10 An abduction-adduction leg tremor is more prevalent in LRRK2-associated disease.18 The most common pathological features are Lewy bodies with tau and ubiquitin staining being less common. Symptomatic treatment is typically required later in the disease course and patients are less prone to dyskinesia.18 Patients with the G2019S mutation are more prone to dystonia in the first two years.18 Penetrance increases with age with a risk estimation of 28% at 59 years and 74% at 79 years.15
- VPS35 (Vacuolar sorting protein 35), OMIM: 601501
In 2011 a mutation p.Asp620Asn (D620N) in VPS35 gene was described12 in familial and rarely in sporadic PD. It is the only pathogenic VPS35 mutation found to date and was the first PD-related pathogenic mutation discovered using next generation sequencing. The phenotype resembles sporadic PD with an earlier age-of-onset at 60 years, rare dementia and a good response to levodopa12 (Table 1). Depression is more common.19 Pathological features include gliosis, tau and alpha-synuclein inclusions.
- EIF4G1 (Eukaryotic translation initiation factor 4 gamma 1), OMIM: 600495.
In 2011 mutations in EIF4G1 were reported in monogenic and sporadic PD.20 The only confirmed mutation associated with PD is A1205H, described in French families and one Irish patient (Table 1) who is a 66-year old Irish man with levodopa-responsive parkinsonism. He developed dyskinesia 15 years after onset treated successfully with deep brain stimulation. He later developed visual hallucinations and dementia 29 years after onset17 There is inconclusive evidence as to whether other EIF4G1 mutations could be pathological.6
- DCTN1 (Dynactin 1) mutations in Perry syndrome, OMIM: 601143
While rare, the syndrome has a very characteristic clinical profile of early-onset parkinsonism, depression, severe weight loss and hypoventilation which can culminate in respiratory failure21 (Table 1). Eight autosomal dominant mutations described in 16 families are causative.22 Patients typically present in the fifth or sixth decades, with mild parkinsonism and a moderate response to levodopa. Patients succumb to respiratory failure; early diagnosis may improve quality of life and potentially offset episodes of respiratory failure with the use of nocturnal bipap or a diaphragmatic pacemaker (Figure 1).
- Rapid onset dystonia parkinsonism / DYT12, OMIM: 182350
The rapid onset of dystonia over a period of days to weeks, frequently after times of stress with associated dysphagia, dysphonia and parkinsonism in the second and third decades of life are the cardinal clinical features of this autosomal dominant disorder (dystonia more prominent than parkinsonism)23,24 (Table 1). Non-motor symptoms such as psychosis, depression and anxiety may be present. Mutations in the ATP1A3 gene are causative. More recently mutations in this gene have been associated with alternating hemiplegia of childhood with early-onset dystonia (<18 months), developmental delay and fluctuating consciousness.23
Autosomal recessive forms:
Some of the unique characteristics, with some overlap, associated with Parkin, PINK1 and DJ-1 are described below5 (Figure 1).
- PARKIN (PARK2), OMIM: 602544.
Parkin was the first autosomal recessive gene linked to PD.5 It encodes a 465 amino-acid protein, the second largest gene in the human genome.5 Parkin is responsible for 50% of autosomal recessive monogenic PD and 15% of the sporadic early-onset (<45) PD.12 Parkin-related disease has a mean age-of-onset of 36 years (associated with homozygous and compound heterozygous mutations)25 and an excellent response to levodopa therapy.26,27 Over 200 mutations have been described (up to May 2015).28 The phenotype is characterised by early dyskinesia in feet and legs,29 motor fluctuations, symmetric onset, hyperreflexia, frequent dystonia6 and a slow disease course without dementia (Table 1).30 The lowest effective levodopa dose should be prescribed. The recognition and diagnosis of parkin-related disease has implications for both treatment and prognosis in these younger patients. Diurnal variation with sleep benefit occurs.9 Lewy bodies are not present.26,27 Parkin can be thought of as a ‘nigropathy’, a less diffuse process without the development of cognitive impairment or anosmia.27,31 Pathology is predominantly restricted to brainstem without Lewy bodies similar to that found in the “frozen addicts” post MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine).
- PINK1 (PTEN- induced putative kinase 1), (PARK6), OMIM: 608309
PINK1 is a 581 amino-acid protein kinase with over 60 mutations reported.5 Mutations in PINK1 are responsible for 2-4% early-onset monogenic PD in Caucasians and 4-9% in Asians.6 Similarly to the phenotype associated with Parkin mutations disease progression is slow and response to levodopa is good. Sense of smell is less likely to be affected than in sporadic PD.31 Psychiatric co-morbidity and gait disturbance is common in PINK1 compared to Parkin (Table 1, Figure 1).32,33 The one reported post-mortem of a patient with compound heterozygote mutation showed substantia nigra pars compacta neuronal loss, Lewy bodies and aberrant neuritis in brainstem, pars compacta and nucleus of Meynert with sparing of the amygdala and locus ceruleus.32
- DJ-1 (PARK 7), OMIM: 602533
DJ-1 occurs in 1-2% of an early-onset of monogenic PD and there are 10 mutations described to date. The phenotype is similar to that of Parkin with an early-onset disease between the ages 20 and 40, good levodopa response and slow progression (Table 1, Figure 1).6 Psychiatric (cognitive impairment, anxiety and depression) features are also reported.6
Genes associated with juvenile onset, atypical forms of parkinsonism:
- ATP13A2 (ATPase type 13A2), (PARK9) (Kufor-Rakeb syndrome), OMIM: 610513
ATP13A2 is a large gene encoding a 1180 amino-acid protein with 10 known pathogenic mutations.5 Kufor-Rakeb syndrome is named after a village in Jordan, where the disease was first described in 1994.33 The phenotype is one of very early-onset disease between the ages of 11 and 16 years, recessive pattern of inheritance, rapid progression, atypical features, pyramidal signs, supranuclear gaze palsy5 and dementia, and facial-faucal-finger minimyoclonus (Table 1).6,12 Recent reports describe more typical early-onset PD.13
- PLA2G6 (Phospholipase A2, group 6) (PARK14), OMIM: 603604.
PLA2G6 displays recessive inheritance, with disease onset ranging from infantile to early adulthood. In infants the onset is before the age of two years35 and is associated with spasticity in all limbs, cognitive decline, bulbar dysfunction, dystonia and cerebellar ataxia. Homozygous mutations in PLA2G6 can occur in adults with a levodopa-responsive dystonia-parkinsonism, pyramidal signs, cognitive dysfunction, Lewy body disease and sometimes iron accumulation in the brain (table 1).6,12,36 There is a debate as to whether heterozygote “carriers” manifest the disease. For example a 28-year-old woman had a single heterozygote (c.238 G>A p.A80T) mutation in PLAG2G6 with a phenotype in keeping with PARK14. She had difficulty speaking, marked tremor, bradykinesia, rigidity, ataxia and anxiety at the age of 18. She developed laterocollis, retrocollis, hand dystonia, dysarthria, dysphonia, limited vertical and horizontal gaze, freezing episodes, marked on dyskinesia and suboptimal response to levodopa. There was a late-onset parkinsonism with dystonia in two paternal grand aunts.
- FBXO7 (F-box protein 7) (PARK 15), OMIM: 605648
FBXO7 encodes a 522 amino-acid protein with four known mutations causing juvenile, progressive, recessive levodopa responsive Parkinsonism. Pyramidal signs and early dystonia also occur (Table 1).37,38
Genetic risk factors:
- GBA, OMIM: 606463.
Recessive mutations in GBA (glucocerebrosidase) gene cause Gaucher’s disease, while heterozygote mutations increase by five fold the risk of developing late-onset PD.39 Mutations in GBA are more prevalent in Ashkenazi Jews (19.6% in comparison to 6.9% of non-Ashkenazi Jewish patients in one study).5 The phenotype is levodopa-responsive parkinsonism with a slightly earlier age-of-onset,12 early motor complications and higher prevalence of dementia.40
- LRRK2 risk factors and protective loci
Ross et al. carried out an assessment of 121 exonic LRRK2 variants in 8,611 patients and 6,929 controls from Caucasian, Asian and Arab Berber populations.41 Carriers of known mutations were excluded and risk associations were described for polymorphisms in Caucasians (p.M1646T) and Asians (p.A419V). Risk association in Asians with p.G2385R was confirmed, while no association was described for p.R1628P. Lastly P.Y2189C could possibly be a risk factor in the Arab-Berber population.41
Ross et al. also identified a common three-variant haplotype (N551K-R1398H-K1423K) that seemed to act in a protective manner and suggested that the reduced penetrance found in LRRK2-associated monogenic PD might be due to variants acting in cis or trans with the pathogenic variant. It is possible that LRRK2 activity influences symptom onset and any future therapeutic suggestions that lower risk in LRRK2 associated monogenic PD might protect against symptomatic onset in sporadic PD. For example the protective R1398H variant has reduced kinase activity suggesting this Roc domain substitution might be the most likely functional allele on the haplotype.41
Conclusion
While PD is a complex disorder, certain clinical features combined with age-of-onset may guide the clinician as to which gene should be tested. The clinician should decide on a pattern of inheritance guided by a family pedigree first. When PD occurs in every generation, with one parent or 50% of children affected, it suggests an autosomal dominant pattern. In autosomal recessive inheritance the disease skips generations, parents are not affected (carrier state) and only 25% of the siblings have PD (although reduced penetrance in autosomal dominant inheritance may mimic recessive disease).5 Secondly, it is important to establish the age-of-onset of PD and associated clinical features. In general, late onset PD is associated with autosomal dominant forms (except SNCA triplications) with prominent tremor or tremor involving the legs suggesting LRRK2 (adduction-abduction leg tremor)18,40 and lack of tremor associated with SNCA-related disease (Figure 1). Information about the ethnicity of the patient may be useful. For example an autosomal dominant picture in a patient originating from the Middle East, of Jewish ancestry or from North Africa is highly suggestive of the G2019S LRRK2 mutation. Patients with late forms of the disease associated with rapid progression, weight loss and respiratory failure should prompt screening of the DCTN1 gene associated with Perry syndrome (Figure 1). Early onset PD is associated with autosomal recessive forms and SNCA triplications (Figure 1). While the patient with early onset PD with dementia and rapid progression should be tested for SNCA triplications, patients with dementia but slow progression should be screened for PINK1 (Figure 1). Patients with early-onset monogenic and sporadic PD and normal cognition, slowly progressive disease, good levodopa response, early dystonia or leg dystonia should have PARKIN screening performed. If Parkin testing is negative or dystonia is not a feature then DJ1 screening should be considered. Juvenile-onset disease associated with atypical features can often point toward a specific gene. Early-onset parkinsonism with supranuclear palsy, pyramidal signs and dementia is characteristic for mutations in ATP13A2 and PLA2G6 (Figure 1). Suggested approach to the genetic testing in a PD patient is proposed in Figure 1.
Parkinson’s disease is responsible for a growing burden on society and health care services. As more genetic associations are described for this complex disorder, more potential therapeutic targets will emerge. While no drugs have yet come to market based upon genetic data, new insights are uncovering the molecular pathways involved in disease pathogenesis. Future therapies may only benefit patients with a particular genetic profile and certain side effects may be predicted in others. The advent of cheaper next generation sequencing technology will lead to even further associations and risk loci being uncovered. Large scale clinical studies to tease out genotype-phenotype correlations may lead to genomic ‘risk’ profiles. For example, individuals harbouring SNCA duplications may benefit from knockdown therapy while those with LRRK2 mutations may benefit from a kinase inhibitor, probably with treatment commencing in the pre-disease ‘at risk’ state. However, a much clearer understanding of the biological pathways involved will be needed to ensure that the correct molecular targets are identified while minimising the potential side effects.
References
- European Parkinson’s disease association, EPDA, http://www.epda.eu.com/en/about-the-epda/
- Parkinson’s Association of Ireland, Strategic Plan 2010-2013,http://www.parkinsons.ie/userfiles/file/StrategicPlan.pdf
- Olesena J, Gustavssonb A, Svenssond M et al. The economic cost of brain disorders in Europe. European Journal of Neurology. 2012;19:155-62.
- Dorsey ER1, Constantinescu R, Thompson JP et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. 2007. Jan 30;68(5):384-6.
- Klein C, Westenberger A. Genetics of Parkinson’s Disease. Cold Spring Harb Perspect Med. Jan 2012;2(1):a008888.
- Schulte C, Gasser T. Genetic basis of Parkinson’s disease: inheritance, penetrance, and expression. The Application of Clinical Genetics. 2011;4:67-80.
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 27 June 1997;Vol. 276 no. 5321:2045-7.
- Shulman JM, De Jager PL, Feany MB. Parkinson’s disease: genetics and pathogenesis, Annu Rev Pathol. 2011;6:193-222.
- Mori H, Hattori N, Mizuno Y. Genotype-phenotype correlation: Familial Parkinson’s disease. Neuropathology 2003;23:90-94.
- Puschnann A, Wszolek ZK. Genotype-phenotype correlations in Parkinson disease. Movement disorders: Genetics and Models. Chapter 16:259-72. Edited by LeDoux MS.
- Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D. Alpha-Synuclein p.H50Q, a Novel Pathogenic Mutation for Parkinson’s Disease. Mov Disord. 2013;28:811-813.
- Bonifati V. Genetics of Parkinson’s disease-state of the art 2013. Parkinsonism and Relat Dis. 2014;20S1,S23-S28.
- Lesagne S, Brice A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Gen. 2009;Vol. 18, Review Issue 1, R48-R59.
- Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL et al. A novel α-synuclein missense mutation in Parkinson disease. 2013;80(11):1062-4.
- Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;Jul;7(7):583-90.
- Gosal D, Ross OA, Wiley J, Irvine GB, Johnston JA, Toft M et al. Clinical traits of LRRK2-associated Parkinson’s disease in Ireland: a link between familial and idiopathic PD. Parkinsonism Relat. Disord. 2005;a 11:349-52.
- McCarthy A. Genetic Investigation of Familial, Early-Onset and Sporadic Parkinson’s Disease in Ireland. MD thesis. University College Dublin.
- Healy DG, Wood NW, Schapira AH et al. Test for LRRK2 mutations in patients with Parkinson’s disease. Pract Neurol. 2008;Dec;8(6):381-5.
- Struhal W, Presslauer S, Spielberger S, Zimprich A, Auff E, Bruecke T et al. VPS35 Parkinson’s disease phenotype resembles the sporadic disease. J Neural Transm. 2014;Jul;121(7):755-9.
- Chartier-Harlin MC, Dachsel JC, Vilariño-Güell C, Lincoln SJ, Leprêtre F, Hulihan MM et al. Translation Initiator EIF4G1 Mutations in Familial Parkinson Disease. Am J Hum Genet. 2011;Sep 9;89(3):398–406.
- Farrer MJ, Hulihan MM, Kachergus JM, Dächsel JC, Stoessl AJ, Grantier LL et al. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;Feb;41(2):163-5.
- Tacik P, Fiesel FC, Fujioka S, Ross OA, Pretelt F, Cardona CC et al. Three families with Perry syndrome from distinct parts of the world. Parkinsonism and Related Disorders. 2014;Volume 20, Issue 8:884–888.
- Cook JF, Hill DF, Snively BM, Boggs N, Suerken CK, Haq I et al. Cognitive impairment in rapid-onset dystonia-parkinsonism. Mov Disord. 2014;Mar;29(3):344-50.
- Dobyns WB, Ozelius LJ, Kramer PL, Brashear A, Farlow MR, Perry TR et al. Rapid-onset dystonia-parkinsonism. 1993;Dec;43(12):2596-602.
- Sun M, Latourelle JC et al. Influence of Heterozygosity for Parkin Mutation on Onset Age in Familial Parkinson Disease, The Gene PD Study. Arch Neurol. 2006;63(6):826-832.
- Polymeropoulos MH1, Higgins JJ, Golbe LI et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. 1996;Nov 15;274(5290):1197-9.
- Doherty KM, Silveira-Moriyama L, Parkkinen L et al. Parkin Disease: A Clinicopathologic Entity? JAMA Neurol. 2013;May;70(5):571-9.
- Genetics Home Reference PARK2, http://ghr.nlm.nih.gov/gene/PARK2
- Chang FC, Mehta P, Koentjoro B, Latt M, Blair N, Nicholson G et al. Dancing Feet Dyskinesias: A Clue to Parkin Gene Mutations. Movement Disorders. 2012;27:587-8.
- Lohmann E, Periquet M, Bonifati V, Wood NW, De Michele G, Bonnet AM, Fraix V, How much phenotypic variation can be attributed to parkin genotype? Annals of Neurology. 2003;54, Issue 2:176-85.
- Verbaan D, Boesveldt S, van Rooden SM, Visser M, Marinus J, Macedo MG et al. Is olfactory impairment in Parkinson disease related to phenotypic or genotypic characteristics? 2008;Dec 2;71(23):1877-82.
- Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer, I, Lorenzo E, Irigoyen J et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain J. Neurol. 2010;133:1128-42.
- Ephraty L, Porat O, Israeli D, Cohen OS, Tunkel O, Yael S et al. Neuropsychiatric and cognitive features in autosomal-recessive early parkinsonism due to PINK1 mutations. Movement Disorders. 2007:22, Issue 4:566-9.
- Najim Al-Din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurologica Scandinavica. 1994;Vol 89, Issue 5:347-52.
- Kurian, MA, Morgan NV, MacPherson L, Foster K, Peake D, Gupta R et al. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). 2008;70:1623-9.
- Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. 2010;Oct 12;75(15):1356-61.
- Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. 2009;Jan 20;72(3):240-5.
- Davison C. Pallido-pyramidal disease. J Neuropathol Exp Neurol. 1954;Jan;13(1):50-9.
- Gan-Or Z, Giladi N, Orr-Urtreger A. Differential phenotype in Parkinson’s disease patients with severe versus mild GBA mutations. 2009;Oct;132(Pt 10):e125.
- Angeli A, Mencacci NE, Duran R, Aviles-Olmos I, Kefalopoulou Z, Candelario J et al. Genotype and phenotype in Parkinson’s disease: lessons in heterogeneity from deep brain stimulation. Disord. Journal 2013;28:1370-5.
- Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G et al. Association of LRRK2 exonic variants with susceptibility to Parkinson‟s disease: a case-control study. Lancet Neurol. 2011;10:898-908.