
Abstract
Huntington’s disease (HD) is an autosomal dominant, progressive, neurodegenerative disorder; most commonly affecting adults.1 A mutant expansion of a CAG repeat >36 copies on chromosome 4, coding for the Huntingtin protein (HTT), underlies numerous downstream cellular pathologies. Using murine models that carry the mHTT gene, anti-sense oligonucleotides (ASOs) have been identified that can reduce the progression of signs in affected mice; one of which has now been translated into human trials. Many researchers maintain that the huge variation between human and murine genomes, behaviour and complexity means that treatments cannot be readily translated between the two species. Many similarities have been found between the nucleotide structure of murine and human HTT genes, allowing for both viable transgenic mice and translatable RNA therapies to be developed.
A mutant expansion of a CAG repeat >36 copies on chromosome 4, coding for the abnormal Huntingtin protein (HTT), leads to numerous downstream cellular problems; culminating in cell death including the striatal medium spiny neurons.2 Development of murine HD models has allowed for the investigation of RNA targeting treatments,3 that have subsequently been the subject of both primate and human safety trials. Use of these models has facilitated advances in the development of both allele non-specific and allele specific approaches to gene suppression, including the first human trial.4 This review discusses current RNA treatments in HD and the issues that complicate their translation from animal models to human trials.
This review is a summary of present knowledge and does not set out to evaluate efficacy of potential treatments, and therefore the specific methodology of animal research is not evaluated here. However, studies using animal models of Huntingtin mRNA suppression with salient results for human application are outlined; followed by an evaluation of murine models of HD and the issues each presents with translating treatment options into humans.
1. Method
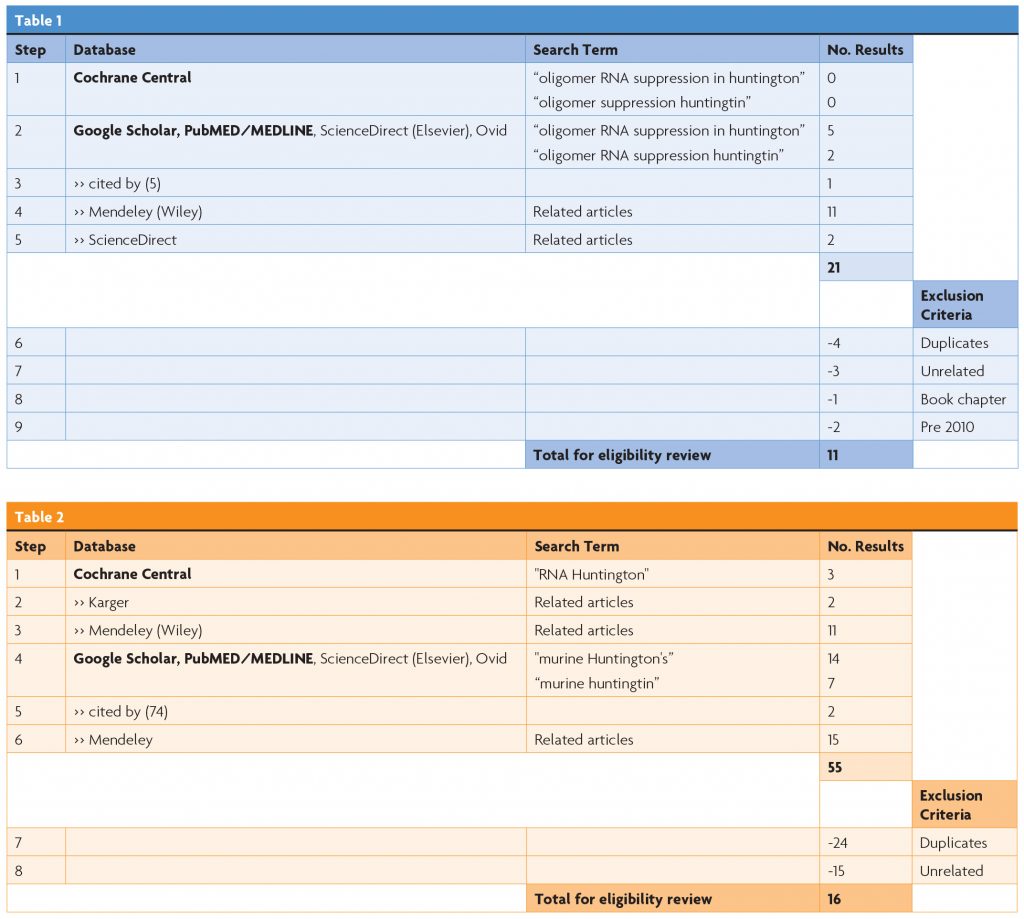
To source trials for this literature review, Cochrane CENTRAL and Google Scholar were independently accessed, and a computerised search was undertaken. Results were processed in line with PRISMA guidelines.5
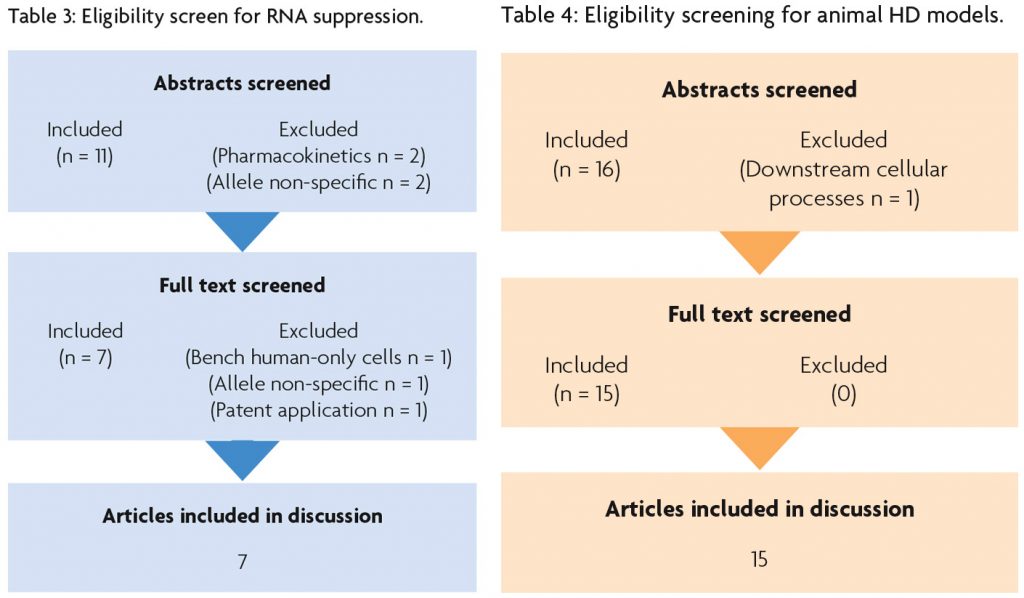
Table 1 describes the search protocol for RNA suppression and Table 2 describes the search protocol for animal models of HD.
2. Results
3. RNA Suppression
The disease phenotype of HD is generated by toxic accumulation of mutant HTT (mHTT) in neurons including the medium spiny neurons of the striatum, as well as accompanying glial cells. This mutant protein has a defective tertiary structure due to excessive replication (>36) of a cytosine-adenine-guanine (CAG) sequence within exon 1 of the gene.2 Due to the autosomal dominant nature of HD, patients also have an unaffected allele which codes for wild-type HTT (wtHTT).
RNA suppression works by introducing short strands of nucleotides that complement segments of mRNA coded for by a gene; binding to them and, through one of several mechanisms, preventing them from being translated into protein. These strands are called antisense oligonucleotides (ASO). Many disorders can be treated using SC or IV introduction of water-soluble oligonucleotides; using sugar backbones as a vector for transport into the cell.6 Oral administration of second generation ASOs is also possible utilising permeation enhancers such as sodium caprate, with acceptable resultant levels of plasma bioavailability.7 However, the resulting molecules are often incapable of being transported across the blood brain barrier; so for HD, short stranded oligonucleotides are injected directly into the CSF of the affected subject where they are taken up by neural cells.6
To study the potential use of ASOs in HD, murine models of HD were used which were first generated shortly after the gene was discovered in the mid-1990s. There are three main types of murine HD homolog: Transgenic mice, knock-in mice and YAC/BAC mice.2 Transgenic mice carry two natural copies of murine wtHTT gene, in addition to the inserted copy of exon 1 of human mHTT gene on a random locus of their genome; thus producing both wtHTT and mHTT as well as displaying an HD phenotype.3 Knock-in mice have the expanded CAG repeat inserted into their existing murine HTT gene and so produce only mHTT.3 Lastly, YAC/BAC mice have the full human HTT gene introduced into their cells via yeast or bacterial DNA and so, like transgenic mice, also produce both forms of HTT.3
Historically, RNA suppression treatments have focused on inhibiting both wtHTT and mHTT production using nucleotide sequences from exons on the 5’ end of resulting mRNA. Using phosphorothioate modified oligonucleotides that catalyse RNase degradation of HTT mRNA, this has been demonstrated to be effective in reducing abnormal signs in HD transgenic mice; not only during treatment but for an extended period after final administration of the ASO.8 Currently the ASO identified by Kordasiewicz and colleagues is the only HTT-suppressing drug to have completed stage 1 human trials and is currently being developed for bigger phase II clinical trials (please see section 4 for further discussion).
However, wtHTT is essential for normal neural development and health, including in CREB (and therefore BDNF) production, mitochondrial energy conversion and other essential cell processes.9,10 More recent research has therefore focused on identifying potential ASOs that are allele specific to mHTT.11
Gagnon et al (2010)12, Skotte et al (2014),13 Rue et al (2016)14 and Datson et al (2017)15 all identified potentially therapeutic, allele specific, locked nucleic acid-modified ASOs (LNA-ASO), targeting the mutant length CAG repeats in mHTT mRNA; whilst sparing both wtHTT and other CAG repeat-containing genes in transgenic mice. These ASOs also did not catalyse RNase and furthermore multiple ASO strands worked in conjunction on very large CAG repeats, indicating disruption of translation as the mechanism of action.12,14 Likewise; Carroll et al13 identified LNA-ASOs capable of selectively binding to SNPs on segments of mHTT mRNA in YAC/BAC mice. In each case, administration displayed benefits in terms of the progression of signs in these transgenic, knock-in or YAC/BAC mice.
Miniarikova et al (2016)16 created four expression cassette-optimised artificial microRNAs (miRNA) targeting human HTT exon 1 (miH), the expanded CAG repeats (miCAG), C or T isoform of SNP rs362331 in exon 50 (miSNP50C and miSNP50T) and the T isoform of SNP rs362307 in exon 67 extended to 3’UTR (miSNP67T). Complete silencing of wtHTT and mHTT was achieved targeting exon 1 and miCAG. mHTT specific silencing was achieved targeting the heterozygous SNP rs362331 in exon 50 or rs362307 in exon 67.16
Sun et al (2014)17 identified a phosphorodiamidate morpholino oligonucleotide (PMO) capable of selectively suppressing mHTT mRNA. PMOs have the benefit of being a particularly stable, soluble and non-toxic type of ASO. The study found CAG repeat-targeting PMOs, with one strand being able to suppress mHTT in both transgenic and knock-in mice.17
4. Translating animal HD models to humans
Each type of murine HD model comes with its own issues in terms of how the results from them can be translated to clinical studies.
Transgenic mice such as R6/1 and R6/2 have a tendency to show severe signs consistent with an HD phenotype early in life.18 This allows for good comparison to juvenile HD as well as rapid screening of treatments for clinical effects. Likewise, YAC mice show early cellular changes reflecting those of human HD.19 Whilst both types of mice show the characteristic progressive phenotype, the issue remains that they do not represent a direct genetic correspondent of human HTT production.3 The addition of human mHTT alleles, or segments thereof, into a random locus of the murine genome means HTT mRNA is not being produced by this gene in its natural locus. In R6/2 mice, the addition of the transgene causes a coincident deletion of the Gm12695 gene; causing expression of a partial fragment. Such expressions displayed by this model cause changes in a range of cell processes such as synaptic transmission and cell signalling.20 However, we are not yet fully aware of all linkage relationships with HTT and consequent protein production or downstream mediation in either mice or humans, which poses a potentially major issue in terms of efficacy and safety. In addition, the presence of the human HTT gene may interfere in a linkage manner with the expression of other unrelated murine genes.2
Knock-in mice such as HdhQ9221 or CAG140/15022 more closely resemble naturally occurring HD in humans, as segments of human mHTT gene are inserted into the existing locus of the murine HTT gene. This means that the mice display a genotype with one mHTT gene and one wtHTT gene, like most human subjects.23 Whilst this allows for a more natural development of cell pathology, behavioural correlates are less pronounced and variation in mortality is not reliably useful as a measure for research trials.2,3
In a broader sense, many researchers maintain that the huge variation between human and murine genomes, behaviour and complexity means that treatments cannot be readily translated between the two species. Certainly, the effects of introducing a CAG repeat that is much longer than the one naturally occurring in mice does not necessarily create the same changes seen in humans. In one study, an increase in CAG repeat to a super-long size created a paradoxical increase in life expectancy of affected mice, directly contradictory to the human phenotype.24 However, whilst these are all valid criticisms, many similarities have been found between the nucleotide structure of murine and human HTT genes,25 allowing for translatable RNA strands to be developed.
Trials in more complex animals, with genomes and characteristics that more closely resemble humans, is essential before human trials can be conducted but brings with it issues of cost and numbers of animals that can, and should, be treated pre-clinically.
To date, this has been attempted through the development of both ovine and porcine HD models. Transgenic sheep injected with full-length human mHTT gene containing 73 CAG repeats have shown expression of mHTT and abnormalities in medium spiny neurons. However, these transgenic sheep show no overt phenotype at later stages of development.26
In pigs, trinucleotide repeat lengths more closely resemble those of humans; with CAG repeats of up to 21 identified in the porcine HTT allele.27 Minipigs have been identified as a suitable transgenic HD model, as their physiology resembles that of humans in several respects. They have a large brain suitable for imaging and there is 96% homology between porcine and human HTT genes. Similar to ovine models, transgenic pigs have historically showed abnormalities of striatal structures, but did not display any behavioural correlates or overt phenotype.28 However, progress with porcine models has been made recently with a knock-in porcine HD model that displays consistent abnormalities reflective of human HD.29 These pigs also show selective degeneration of striatal medium spiny neurons and the incorporated CAG repeat extension is germline transmissable. This model represents the most analogous model of human HD in a large mammal to date and provides a promising platform for treatment trials.
Likewise, no naturally occurring HD homolog exists in non-human primates. Currently, trials to establish the safety of reducing wtHTT in adult primates (in line with the Kordasiewicz (2012) study)8 have been conducted and the impact of ASO reduction of wtHTT was found to have no adverse effects; with the exception of a temporary loss of knee reflexes in monkeys, which may be more associated with the mode of delivery than the therapeutic agent itself.30 It may soon be possible to emulate HD in non-human primates via one of two methods: hyperexpression of mHTT activated by viral vectors (Ramaswamy et al., 2007),2 or using transgenic rhesus macaques carrying the mutant exon-1 of human IT15 (Yang et al., 2008).31
Whilst the treatment option currently being studied in a human trial (IONIS HTTRx) developed from the findings of Kordasiewicz and colleagues (2012)10 provides a promising option for adults with HD, with no adverse reported events to date, it’s lack of specificity for mHTT does mean it may need modifying or an alternate, allele specific ASO developed. This is particularly true for juvenile HD, in which inhibition of wtHTT may have significant (and incompletely known) effects for patients. Given this, one would predict that a focus on allele specific PMOs or LNA-ASOs will eventually take precedence in HD research. Genetic variability and further understanding of the different genotypes and phenotypes of HD will inevitably provide avenues for treatment development, such as that of Skotte and colleagues (2014);13 but also complications for developing mHTT specific ASOs effective in all HD variants.
Conclusion
RNA suppression works through the introduction of ASOs into the CSF that either inhibit HTT mRNA translation or catalyse HTT mRNA degradation. This process can be allele specific to mHTT mRNA, or non-specific affecting both mHTT and wtHTT production. Using murine models that carry the mHTT gene, ASOs have been identified that are able to reduce signs in affected mice; and one of these therapies has now been the subject of a clinical trial in patients with HD. Issues exist when translating findings from murine models to human patients. Work in primate and other large animal models of HD may gain greater prominence in the years to come; with promising models having emerged last year.
References
- Moore D, Puri, B. (2012). Textbook of clinical neuropsychiatry and behavioral neuroscience. Hodder Education. Retrieved from https://bit.ly/2Id7ygr
- Ramaswamy S, McBride JL, Kordower JH. Animal Models of Huntington’s Disease. ILAR Journal. 2007;48(4):356–373. https://doi.org/10.1093/ilar.48.4.356
- Ferrante RJ. Mouse models of Huntington’s disease and methodological considerations for therapeutic trials. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease. 2009;1792(6):506–520. https://doi.org/10.1016/j.bbadis.2009.04.001
- Keiser MS, Kordasiewicz HB, McBride JL. Gene suppression strategies for dominantly inherited neurodegenerative diseases: lessons from Huntington’s disease and spinocerebellar ataxia. Human Molecular Genetics. 2016;25(R1):R53-64. https://doi.org/10.1093/hmg/ddv442
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Medicine, 2009;6(7), e1000097.
- Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Advanced Drug Delivery Reviews. 2015;87:46–51. https://doi.org/10.1016/J.ADDR.2015.01.008
- Tillman LG, Geary RS, Hardee GE. Oral Delivery of Antisense Oligonucleotides in Man. Pharmacists Association J Pharm Sci. 2008;97:225–236. https://doi.org/10.1002/jps.21084
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Cleveland DW. Sustained Therapeutic Reversal of Huntington’s Disease by Transient Repression of Huntingtin Synthesis. Neuron. 2012;74(6):1031–1044. https://doi.org/10.1016/J.NEURON.2012.05.009
- White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nature Genetics. 1997;17(4):404–410. https://doi.org/10.1038/ng1297-404
- Zuccato C. Loss of Huntingtin-Mediated BDNF Gene Transcription in Huntington’s Disease. Science. 2001;293(5529):493–498. https://doi.org/10.1126/science.1059581
- Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, Hayden MR. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin. Molecular Therapy : The Journal of the American Society of Gene Therapy. 2011;19(12):2178–2185. https://doi.org/10.1038/mt.2011.201
- Gagnon KT, Pendergraff HM, Deleavey GF, Swayze EE, Potier P, Randolph J, Corey DR. (2010). Allele-selective inhibition of mutant huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry. https://doi.org/10.1021/bi101208k
- Skotte NH, Southwell AL, Østergaard ME, Carroll JB, Warby SC, Doty CN, Hayden MR. Allele-specific suppression of mutant huntingtin using antisense oligonucleotides: providing a therapeutic option for all Huntington disease patients. PloS One, 2014;9(9), e107434. https://doi.org/10.1371/journal.pone.0107434
- Rué L, Bañez-Coronel M, Creus-Muncunill J, Giralt A, Alcalá-Vida R, Mentxaka G, Martí E. Targeting CAG repeat RNAs reduces Huntington’s disease phenotype independently of huntingtin levels. The Journal of Clinical Investigation. 2016;126(11):4319–4330. https://doi.org/10.1172/JCI83185
- Datson NA, González-Barriga A, Kourkouta E, Weij R, van de Giessen J, Mulders S, van Deutekom JCT. The expanded CAG repeat in the huntingtin gene as target for therapeutic RNA modulation throughout the HD mouse brain. PloS One, 2017;12(2), e0171127. https://doi.org/10.1371/journal.pone.0171127
- Miniarikova J, Zanella I, Huseinovic A, van der Zon T, Hanemaaijer E, Martier R, Konstantinova P. (2016). Design, Characterization, and Lead Selection of Therapeutic miRNAs Targeting Huntingtin for Development of Gene Therapy for Huntington’s Disease. Molecular Therapy. Nucleic Acids. 5, e297. https://doi.org/10.1038/mtna.2016.7
- Sun X, Marque LO, Cordner Z, Pruitt, JL, Bhat M, Li PP, Rudnicki DD. Phosphorodiamidate morpholino oligomers suppress mutant huntingtin expression and attenuate neurotoxicity. Human Molecular Genetics. 2014;23(23):6302–6317. https://doi.org/10.1093/hmg/ddu349
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Bates GP. Exon 1 of the HD Gene with an Expanded CAG Repeat Is Sufficient to Cause a Progressive Neurological Phenotype in Transgenic Mice. Cell. 1996;87(3):493–506. https://doi.org/10.1016/S0092-8674(00)81369-0
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Hayden MR. A YAC Mouse Model for Huntington’s Disease with Full-Length Mutant Huntingtin, Cytoplasmic Toxicity, and Selective Striatal Neurodegeneration. Neuron. 1999;23(1):181–192. https://doi.org/10.1016/S0896-6273(00)80764-3
- Jacobsen JC, Erdin S, Chiang C, Hanscom C, Handley RR, Barker DD, Talkowski ME. (2017). Potential molecular consequences of transgene integration: The R6/2 mouse example OPEN. Nature Publishing Group. https://doi.org/10.1038/srep41120
- Wheeler V, Auerbach W, White JK, Srinidhi J, Auerbach A, Ryan A, MacDonald ME. Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Human Molecular Genetics. 1999;8(1):115–122. https://doi.org/10.1093/hmg/8.1.115
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. The Journal of Comparative Neurology. 2003;465(1):11–26. https://doi.org/10.1002/cne.10776
- Menalled LB. Knock-in mouse models of Huntington’s disease. NeuroRX. 2005;2(3):465–470. https://doi.org/10.1602/neurorx.2.3.465
- Morton AJ, Glynn D, Leavens W, Zheng Z, Faull RLM, Skepper JN, Wight JM. Paradoxical delay in the onset of disease caused by super-long CAG repeat expansions in R6/2 mice. Neurobiology of Disease. 2009;33(3):331–341. https://doi.org/10.1016/j.nbd.2008.11.015
- Lin B, Nasir J, Kalchman MA, Mcdonald H, Zeisler J, Goldberg YP, Hayden MR. Structural analysis of the 5′ region of mouse and human huntington disease genes reveals conservation of putative promoter region and di- and trinucleotide polymorphisms. Genomics. 1995;25(3):707–715. https://doi.org/10.1016/0888-7543(95)80014-D
- Jacobsen JC, Bawden CS, Rudiger SR, McLaughlan CJ, Reid SJ, Waldvogel HJ, Snell RG. An ovine transgenic Huntington’s disease model. Human Molecular Genetics. 2010;19(10):1873–1882. https://doi.org/10.1093/hmg/ddq063
- Madsen LB, Thomsen B, Sølvsten CAE, Bendixen C, Fredholm M, Jørgensen AL, Nielsen AL. (2007). Identification of the porcine homologous of human disease causing trinucleotide repeat sequences. Neurogenetics. https://doi.org/10.1007/s10048-007-0088-y
- Baxa M, Hruska-Plochan M, Juhas S, Vodicka P, Pavlok A, Juhasova J, Motlik J. (2013). A transgenic minipig model of huntington’s disease. Journal of Huntington’s Disease. https://doi.org/10.3233/JHD-130001
- Yan S, Tu Z, Liu Z, Fan N, Yang H, Yang S, Li, XJ. A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell. 2018;173(4):989–1002.e13. https://doi.org/10.1016/J.CELL.2018.03.005
- McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, Davidson BL. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Molecular Therapy: The Journal of the American Society of Gene Therapy. 2011;19(12):2152–2162. https://doi.org/10.1038/mt.2011.219
- Yang SH, Cheng PH, Banta H, Piotrowska-Nitsche K, Yang JJ, Cheng ECH, Chan AWS. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature. 2008;453(7197):921–924. https://doi.org/10.1038/nature06975
Further reading
- Bae B-I, Hara MR, Cascio MB, Wellington C.L, Hayden MR, Ross CA, Sawa A. Mutant huntingtin: nuclear translocation and cytotoxicity mediated by GAPDH. Proceedings of the National Academy of Sciences of the United States of America, 2006;103(9):3405–3409. https://doi.org/10.1073/pnas.0511316103
- Barnes GT, Duyao MP, Ambrose CM, McNeil S, Persichetti F, Srinidhi J, MacDonald ME. Mouse Huntington’s disease gene homolog (Hdh). Somatic Cell and Molecular Genetics, 1994;20(2):87–97. https://doi.org/10.1007/BF02290678
- Baxendale S, Abdulla S, Elgar G, Buck D, Berks M, Micklem G, Lehrach H. Comparative sequence analysis of the human and pufferfish Huntington’s disease genes. Nature Genetics, 1995;10(1):67–76. https://doi.org/10.1038/ng0595-67
- Beal MF, Ferrante RJ. Experimental therapeutics in transgenic mouse models of Huntington’s disease. Nature Reviews. Neuroscience, 2004;5(5):373–384. https://doi.org/10.1038/nrn1386
- Borovecki F, Lovrecic L, Zhou J, Jeong H, Then F, Rosas HD, Krainc D. Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proceedings of the National Academy of Sciences of the United States of America, 2005;102(31):11023–11028. https://doi.org/10.1073/pnas.0504921102
- Buraczynska MJ, van Keuren ML, Buraczynska K, Chang YS, Crombez E, Kurnit DM. Construction of human embryonic cDNA libraries: HD, PKD1 and BRCA1 are transcribed widely during embryogenesis. Cytogenetic and Genome Research, 1995;71(2):197–202. https://doi.org/10.1159/000134106
- Burke JR, Enghild JJ, Martin ME, Jou YS, Myers RM, Roses AD, Strittmatter WJ. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nature Medicine, 1996;2(3):347–350. https://doi.org/10.1038/nm0396-347
- Hu Y, Chopra V, Chopra R, Locascio JJ, Liao Z, Ding H, Scherzer CR. Transcriptional modulator H2A histone family, member Y (H2AFY) marks Huntington disease activity in man and mouse. Proceedings of the National Academy of Sciences of the United States of America, 2011;108(41):17141–17146. https://doi.org/10.1073/pnas.1104409108
- Lin C-H, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Human Molecular Genetics, 2001;10(2):137–144. https://doi.org/10.1093/hmg/10.2.137
- Mandich P, Di Maria E, Bellone E, Ajmar F, Abbruzzese G. Molecular Analysis of the IT15 Gene in Patients with Apparently “Sporadic” Huntington’s Disease. European Neurology, 1996;36(6):348–352. https://doi.org/10.1159/000117292
- Mangiarini L, Sathasivam K, Seller M, Cozens, Harper A, Hetherington C, Bates GP. Exon 1 of the HD Gene with an Expanded CAG Repeat Is Sufficient to Cause a Progressive Neurological Phenotype in Transgenic Mice. Cell, 1996;87(3):493–506. https://doi.org/10.1016/S0092-8674(00)81369-0
- Pouladi MA, Morton AJ, Hayden MR. (n.d.). Choosing an animal model for the study of Huntington’s disease.
- Runne H, Kuhn A, Wild EJ, Pratyaksha W, Kristiansen M, Isaacs JD, Luthi-Carter R. Analysis of potential transcriptomic biomarkers for Huntington’s disease in peripheral blood. Proceedings of the National Academy of Sciences of the United States of America, 2007;104(36):14424–14429. https://doi.org/10.1073/pnas.0703652104
- Sadri-Vakili G, Cha J-HJ. Mechanisms of disease: Histone modifications in Huntington’s disease. Nature Clinical Practice. Neurology, 2006;2(6):330–338. https://doi.org/10.1038/ncpneuro0199
- Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, Guhathakurta D, Lusis AJ. An integrative genomics approach to infer causal associations between gene expression and disease. Nature Genetics, 2005;37(7):710–717. https://doi.org/10.1038/ng1589
- Schoch KM, Miller TM. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron, 2017;94(6):1056–1070. https://doi.org/10.1016/J.NEURON.2017.04.010
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends in Genetics : TIG, 2003;19(5):233–238. https://doi.org/10.1016/S0168-9525(03)00074-X
- von Horsten S, Schmitt I, Nguyen HP, Holzmann C, Schmidt T, Walther T, Riess O. Transgenic rat model of Huntington’s disease. Human Molecular Genetics, 2003;12(6):617–624. https://doi.org/10.1093/hmg/ddg075
- Yamamoto A, Lucas JJ, Hen R. Reversal of Neuropathology and Motor Dysfunction in a Conditional Model of Huntington’s Disease. Cell, 2000;101(1):57–66. https://doi.org/10.1016/S0092-8674(00)80623-6