
Summary
- The first 100 genetic loci affecting MS susceptibility have been identified
- The genes indicate susceptibility is mainly due to variation in immune response
- The gene list includes current targets for MS therapy, and so likely future targets
- The two genes regulating Vitamin D activation are risk genes
- Biomarkers, and new therapeutic targets for existing and novel molecules, should follow from these genetic discoveries.
Multiple sclerosis (MS) is one of the most common neurological diseases of young adults. It is primarily an inflammatory disorder of the brain and spinal cord in which focal lymphocytic infiltration leads to damage of myelin and axons.1 The risk of an individual developing MS increases greatly with the relatedness to someone who has MS. Typically from 1 in a 1000 for the normal population, about 10 fold higher for a first degree relative with MS, and about 200 fold higher for an identical twin with MS.1 This indicates genetic variants can increase MS susceptibility, and identification of the genes should enable a better understanding of MS pathogenesis, development of better therapeutics, discovery of biomarkers to predict and measure response to therapies, and so better clinical management of the disease. Excitingly, many of these genetic variants have now been identified, as described in two recent papers in Nature (2011)2 and Nature Genetics (2013).3 However, it’s notable that if one identical twin has MS, the other most likely will not have MS.1 This suggests it will be unlikely that genetic variants can be used to predict MS risk accurately in the clinic, since even for those identical genetically there is less than a 25% chance of concordance. Environmental factors are clearly also of major importance in disease risk, and these include exposure to UV light, vitamin D levels, smoking, and exposure to Epstein Barr virus.4 Concomitant with the discovery of the genes affecting MS, has been the sudden increase in pharmaceutical options for the most common form of MS, relapsing remitting MS. These new therapies can be more effective than previous therapies in reducing relapse rates, disability, disease progression, and brain damage as assessed by changes in brain volume and increase in gadolinium enhance lesions. But drugs which have significant effects on relapse rate and progression such as alemtuzumab and nataluzimab, carry increased risk of serious side effects. MS progression is highly variable and individuals are most responsive to disease modifying therapies when in the early stages of disease.5 Since individuals are likely to vary in their response to each therapy, the decisions of which therapy to use, and how to measure response, are now critical to MS management. Can the identification of the genetic basis for MS risk be used to advance knowledge of such biomarkers? Can it enable the development of new therapies, including for secondary and primary progressive MS? How useful in the clinic is having a list of genes affecting MS risk?
Risk gene discovery
The first MS risk gene, HLA-DRB1 1501, indicated much about pathogenesis. The protein product of this gene has only one known function, to present antigen to CD4 T cells. So CD4 T cells are important in MS, MS is at least in part an immune disease and therapies targeting this interaction between antigen presenting cells and CD4 T cells may be effective: glatiramer acetate arose out of experiments using peptides designed to bind this MHC class II molecule, and interferon beta is an immune modulator. In 2007 two more genes were identified using a genome wide association study (GWAS),6 and there are now (2014) more than 110 genetic regions (the MS110), plus the MHC region, associated with MS.3 The breakthrough in identification of the MS risk genes was enabled by three major steps forward in human genetics: the sequencing of the human genome (2001), the cataloguing of the common genetic variants in the HapMap project (2005) and the 1000 genome project (2012), and the dramatic drop in the cost of genotyping, from about $50/variant to less than $0.01. This, combined with the collection of DNA samples from thousands of patients with MS and ethnically matched controls, with international collaboration mediated by the International MS Genetics Consortium, allowed the necessary genetic experiments and analysis to be done. Many genes which affect MS risk also affect risk of other autoimmune diseases. The immunochip was designed to accelerate the discovery of the next genes in MS and other autoimmune diseases, by loading a genotyping chip with a high density of SNPS from all the genetic regions known to be associated with more than 10 autoimmune diseases from first phase GWAS. This allowed fine mapping of associations, and genotyping of more than 200,000 individuals, including 20,000 with MS, and replication or not of initial findings. This succeeded in finding another 48 MS risk genes, replicating the original Wellcome Trust GWAS findings, and more precisely identifying the regions of association.
How do the risk variants affect MS pathogenesis?
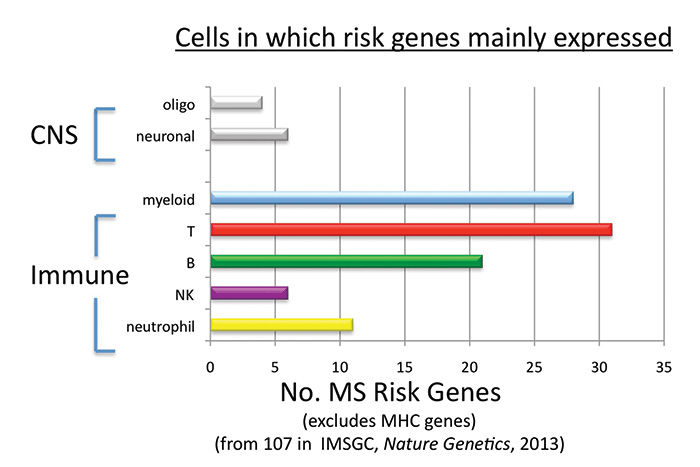
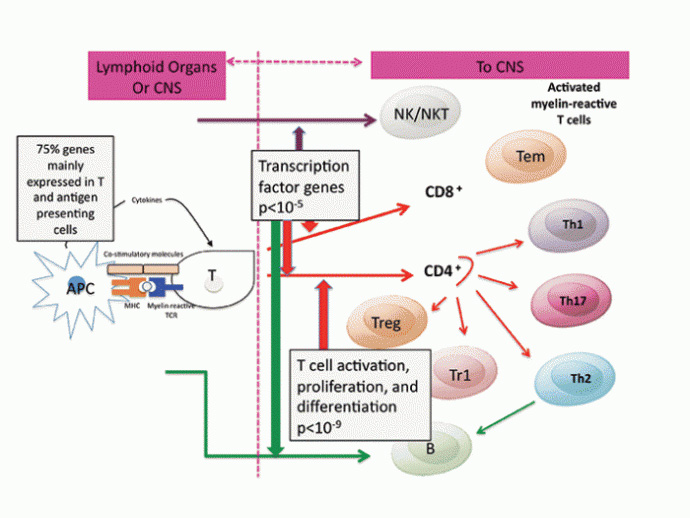
These MS risk genes are mainly expressed in immune cells,7 especially in antigen presenting and T cells, supporting the paradigm that MS is an immunological disease driven by excessive activation of myelin reactive T cells8 (Figures 1 and 2).
Strikingly, most associated SNPs are in intronic or intergenic regions.7 This suggests the genetic variants increase risk by regulating gene expression. The over-representation of transcription factors controlling lymphocyte differentiation, suggests dysregulation of particular immune cell subsets may be driving MS risk, and this information has already been used to identify candidate biomarkers of MS risk, and may prove useful in assessing drug response.9
Do the MS110 indicate therapeutic options?
Proof of principal that the list of genes will be useful for finding new drugs, is that two of the risk genes are targets for existing drugs. IL2R, or CD25, is the target for the monoclonal antibody Daclizumab.10 Natalizumab is a monoclonal antibody to α4-integrin of VLA-4, the CD49d antigen of leukocytes, blocking interaction with its ligand VCAM1, another of the MS risk genes.11 There was also a potential explanation for some therapies working in rheumatoid arthritis but not MS: the genetic variants of CD4012 and TNFRSF1A13 have different associations in the two diseases. The MS110 include many cytokines, their receptors, and other cell surface proteins for which pharma already have libraries of potential ligands. The risk gene products function like levers, the allele state altering the way the lever is pulled, and biological studies have followed to identify how these levers were increasing MS risk, and so whether ligands need to be agonists or antagonists. There is also immediate benefit: amongst the MS110, two genes, CYP27B1 and CYP24A1 regulate vitamin D3 activation. They have no other known function. The protective variant of CYP27B1 is more highly expressed in tolerogenic dendritic cells, which produces more of the tolerising cytokine IL10 in a genotype dependent manner.13 This is the smoking gun showing vitamin D is important in MS pathogenesis, supporting its use in reducing relapse risk,14 and is an example of immediate benefit from finding the MS risk genes.
Are pathogens implicated in MS pathogenesis by the MS110?
If pathogens cause MS or alter its progression appropriate therapies could be employed. Genome wide analysis studies have implicated specific viruses for some autoimmune conditions. Individuals homozygous for the Crohn’s disease risk allele rs601338 of FUT2, the receptor for noroviruses, are protected against norovirus infection, but are more likely to develop Crohn’s and other autoimmune diseases such as type 1 diabetes and inflammatory bowel disease.16 The Epstein Barr virus (EBV) virus has long been implicated as contributing to MS pathology.4 Although its receptors have yet to be associated with MS risk, genetic variants of CD40 are associated with MS, and EBV uses its own analogue of human CD40 to induce infected B cells to proliferate. The genotype with higher expression of CD40 decreases MS risk.15 Since CD40 is a costimulatory molecule, required for T cell activation, it would be expected that higher expression would increase risk of MS. A potential explanation for this paradox is that low host CD40 expression on B cells favours proliferation/survival of EBV infected B cells, using their EBV CD40. Risk genes TNFSF14 and TNFRSF6B may affect entry of herpesvirus 1; and SLAMF7 and TYK2 morbillivirus infection.16 As CD40, TNFSF14/TNFRSF6B and SLAMF7/TYK2 also have roles affecting other aspects of the immune response, further implication of an EBV/HSV1/morbillivirus contribution to MS might follow if the MS susceptibility genotype can be shown to increase tissue damage or other infectious consequences due to these viruses.
What next for MS genetics?
Only a fraction (c.30%) of the heritability of MS has been accounted for.3 The missing heritability could be due to risk variants of smaller effect, rare variants, and epistasis. Such variants are being sorted by increasing sample numbers, and using an MS replication chip which includes all known exonic variants, including rare ones. This will also help refine the regions currently covering large blocks of linkage disequilibrium (where genetic variants are inherited as a block). The risk factors have all been identified by comparing allele frequencies between MS and controls, so they identify susceptibility variants. Such variants must affect pathogenesis, but not necessarily progression. Since therapy choice is dependent on the likely rate of progression, with more conservative choices being favoured for benign MS, a new priority is to identify genetic factors controlling progression. The MS risk genes identified to date provide us with a road map for dissecting out the molecular architecture of MS. So far they indicate MS is an immune cell mediated disease, support a role for vitamin D in pathogenesis and therapy, have already been used to identify candidate biomarkers, and identified new targets for investigation as therapies. This latter is particularly promising, as pharma has already built libraries of compounds to target many of the genes and pathways now known from the gene discoveries to affect MS risk.
References
- Compston A, Coles A. Multiple sclerosis. Lancet 2008;25;372(9648):1502-17.
- International Multiple Sclerosis Genetics Consortium. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214-9.
- International Multiple Sclerosis Genetics Consortium. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013 Nov;45(11):1353-60.
- Pender MP. CD8+ T-Cell Deficiency, Epstein-Barr Virus Infection, Vitamin D Deficiency, and Steps to Autoimmunity: A Unifying Hypothesis. Autoimmune Dis. 2012:189096.
- Trojano M, Pellegrini F, Paolicelli D, Fuiani A, Zimatore GB, Tortorella C, Simone IL, Patti F, Ghezzi A, Zipoli V, Rossi P, Pozzilli C, Salemi G, Lugaresi A, Bergamaschi R, Millefiorini E, Clerico M, Lus G, Vianello M, Avolio C, Cavalla P, Lepore V, Livrea P, Comi G, Amato MP. Italian Multiple Sclerosis Database Network (MSDN) Group. Real-life impact of early interferon beta therapy in relapsing multiple sclerosis. Ann Neurol. 2009 Oct;66(4):513-20.
- International Multiple Sclerosis Genetics Consortium, Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ, Pericak-Vance MA, Gregory SG, Rioux JD, McCauley JL, Haines JL, Barcellos LF, Cree B, Oksenberg JR, Hauser SL. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007 Aug 30;357(9):851-62.
- International Multiple Sclerosis Genetics Consortium. Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. Am J Hum Genet. 2013 Jun 6;92(6):854-65.
- Hafler DA. Perspective: Deconstructing a disease. Nature. 2012 Apr 12;484(7393):S6.
- Parnell GP, Gatt PN, Krupa M, Nickles D, McKay FC, Schibeci SD, Batten M, Baranzini S, Henderson A, Barnett M, Slee M, Vucic S, Stewart GJ, Booth DR. The autoimmune disease-associated transcription factors EOMES and TBX21 are dysregulated in multiple sclerosis and define a molecular subtype of disease. ClinImmunol. 2014 Mar;151(1):16-24.
- Shahijanian F, Parnell GP, McKay FC, Gatt PN, Shojoei M, O’Connor KS, Schibeci SD, Brilot F, Liddle C, Batten M; ANZgene Multiple Sclerosis Genetics Consortium, Stewart GJ, Booth DR. The CYP27B1 variant associated with an increased risk of autoimmune disease is underexpressed in tolerizing dendritic cells. Hum Mol Genet. 2014 Mar 15;23(6):1425-34.
- Bielekova B, Becker BL. Monoclonal antibodies in MS: mechanisms of action. Neurology, 2010;74Suppl 1:S31-40.
- Gandhi KS, McKay FC, Cox M, Riveros C, Armstrong N, Heard RN, Vucic S, Williams DW, Stankovich J, Brown M, Danoy P, Stewart GJ, Broadley S, Moscato P, Lechner-Scott J, Scott RJ, Booth DR; ANZgene Multiple Sclerosis Genetics Consortium. The multiple sclerosis whole blood mRNA transcriptome and genetic associations indicate dysregulation of specific T cell pathways in pathogenesis. Hum Mol Genet. 2010 Jun 1;19(11):2134-43.
- Gregory AP, Dendrou CA, Attfield KE, Haghikia A, Xifara DK, Butter F, Poschmann G, Kaur G, Lambert L, Leach OA, Prömel S, Punwani D, Felce JH, Davis SJ, Gold R, Nielsen FC, Siegel RM, Mann M, Bell JI, McVean G, Fugger L. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature. 2012 Aug 23;488(7412):508-11.
- Ascherio A, Munger KL, Lünemann JD. The initiation and prevention of multiple sclerosis. Nat Rev Neurol. 2012 Nov 5;8(11):602-12.
- Smyth DJ, Cooper JD, Howson JM, Clarke P, Downes K, Mistry T, Stevens H, Walker NM, Todd JA. FUT2 nonsecretor status links type 1 diabetes susceptibility and resistance to infection. Diabetes. 2011 Nov;60(11):3081-4.
- Booth DR. Do pathogens contribute to multiple sclerosis aetiology? Microbiology Australia 2013;3;144-6.