

Summary
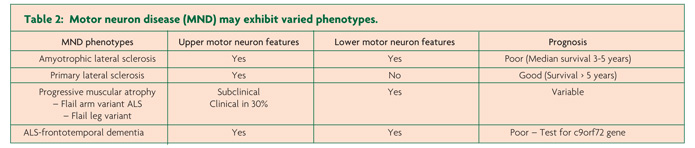
- MND exhibit variable phenotypes
- ALS is the commonest and most lethal of the phenotypes
- Cognitive impairment is a feature of MND
Introduction
Motor neuron disease (MND) encompasses a group of rapidly progressive and universally fatal neurodegenerative disorders of the human motor system, first described in the mid-19th century by the French Neurologist Jean Martin Charcot.1 Amyotrophic lateral sclerosis (ALS) is the commonest MND phenotype, clinically characterised by progressive neurological deterioration and co-existence of upper and lower motor neuron signs.2 In addition, the varied clinical presentations of MND also include (i) progressive muscle atrophy (PMA, ~ 10% of MND cases), a clinically pure lower motor neuron (LMN) phenotype, (ii) primary lateral sclerosis (PLS, 1-3% of MND cases), a clinically pure upper motor neuron (UMN) phenotype and (iii) progressive bulbar palsy (PBP, 1-2% of MND cases), an isolated bulbar phenotype with relative preservation of spinal motor neurons. More recently, an association between ALS and frontotemporal degeneration (FTD) has been established, suggesting that ALS forms a continuum with primary neurodegenerative disorders, a notion underscored by the identification of the c9orf72 hexanucleotide expansion.3,4 Despite the clinical heterogeneity, median survival of MND remains three years, although the atypical phenotypes exhibit a longer survival.5
Amyotrophic lateral sclerosis
In European-based population studies the incidence of ALS appears uniform at 2.16 per 100,000 person-years with a prevalence of 4-6 per 100,000,6 with a lifetime risk of developing ALS being 1 in 400, where the incidence is slightly higher in males [1.2-1.5:1].6 Sporadic ALS peaks between the ages of 50 to 75 years and declines after the age of 80,5 with the age-specific incidence remaining stable over the past decade.7 The frequency of ALS is significantly lower in non-Caucasian populations,8 suggesting a role for genetic factors in ALS susceptibility. A genetic aetiology has been identified in up to 20% of apparently “sporadic” and 60% of familial ALS cases, in which two or more family members are clinically affected, with at least 16 genes and genetic loci implicated in ALS pathogenesis.9
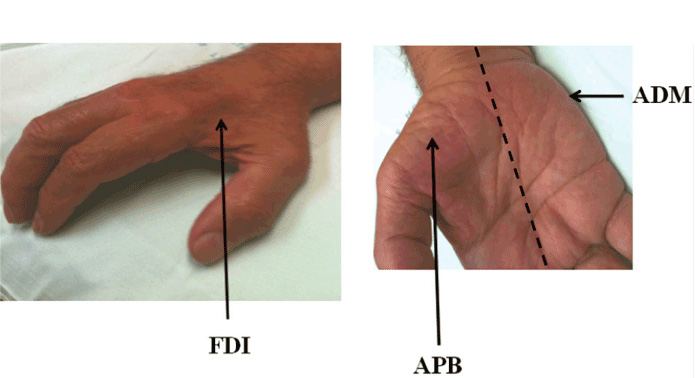
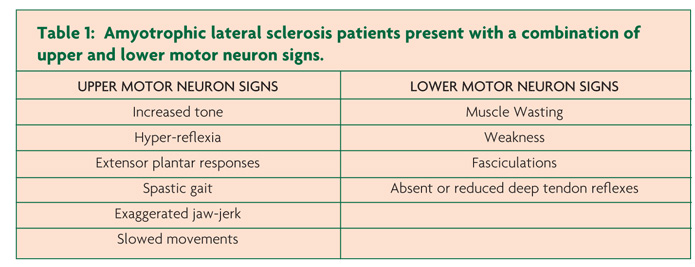
Clinically, ALS is characterised by co-existence of upper and lower motor neuron signs encompassing multiple body regions, with evidence of progressive deterioration.2 Lower motor neuron signs are clinically characterised by fasciculations, muscle wasting and weakness, while UMN signs include slowness of movement, increased tone, hyper-reflexia and extensor plantar responses. The majority of ALS patients present with limb-onset disease (65-75%),10 spreading along the neuraxis to affect contiguous motor neurons.11,12 Preferential wasting and weakness of thenar muscles, termed the split-hand phenomenon (Figure 1), is a specific feature of ALS.13,14 While fasciculations are a cardinal feature of ALS, they are infrequently the presenting symptom.15 Patients presenting solely with fasciculations and muscle cramping should be monitored as these may infrequently progress to develop ALS.16 Extra-ocular and sphincter muscles are preserved until advanced stages of the disease,17 and sensory nerves are not typically affected.5
Bulbar-onset disease may be evident in 20-25% of patients, characterised by progressive dysarthria, dysphagia, hoarseness, tongue wasting, weakness and fasciculations as well as emotional lability.2 Aspiration pneumonia, malnutrition and weight loss are consequent features resulting in an adverse prognosis.18 Respiratory dysfunction is a late feature of ALS, ultimately resulting in terminal respiratory failure,19 although rarely may be the presenting symptom.20,21
The “split hand” sign refers to preferential wasting of the thenar group of muscles, including the abductor pollicis brevis (APB) and first dorsal interosseous (FDI), when compared to the abductor digit minimi (ADM) [Figure 1].14,22 This pattern of muscle atrophy is specific for ALS, and may differentiate ALS from potential mimic disorders.13 The ability to quantify the split hand sign, through the development of a split-hand index (SI), was recently demonstrated to be of diagnostic significance in ALS.23 The mechanisms underlying the split hand in ALS remain elusive, although cortical hyperexcitability seems to be most plausible mechanism.23
In addition to pure motor symptoms, subtle cognitive abnormalities may be evident in up to 50% of ALS patients,24 characterised by executive dysfunction, language and memory impairment along with behavioural abnormalities, which may precede the onset of motor symptoms.24 Recognition of cognitive dysfunction has implication for vital management of ALS, as these symptoms may adversely impact on patient compliance and decision-making abilities. At the extreme end of the spectrum, frontotemporal dementia may develop in up to 15% of ALS patients,6,24 and is clinically characterised by executive and language dysfunction, irrational behavioural, personality changes, apathy, poor insight, loss of empathy, irritability and disinhibition.25
The presence of psychiatric features in the setting of FTD-ALS may be indicative of a recently discovered genetic mutation in the c9orf72 gene on chromosome 9p21.25 Specifically, increased hexanucleotide repeat expansion (GGGGCC) in the intornic segment of the c9orf72 gene, which appears to be dominantly inherited, is causative for both ALS and frontotemporal dementia.3,4 Importantly, the c9orf72 hexanucleotide expansion appeared to underlie over 40% of familial and 20% of sporadic ALS cases in the original studies,3,4 although subsequent studies have established a frequency of 4.1-8.3% in apparently “sporadic” ALS cohorts.26 In addition to predisposition for dementia, the c9orf72 ALS cohorts exhibit an earlier age of onset and shorter survival.27 The c9orf72 discovery has radically altered the understanding of ALS pathogenesis, implying that ALS is a multisystem neurodegenerative disorder, rather than a pure neuromuscular disease.9 Importantly, accumulation of TDP-43 along with p62 positive TDP-43 negative inclusions in hippocampus and cerebellar neurons appears to be neuropathological hallmarks of c9orf72 associated ALS and FTD,28 suggesting the existence of a common pathophysiological pathway, although the precise pathophysiological mechanisms appear to be complex and remain to be fully elucidated.29
The diagnosis of ALS remains clinically based relying on identifying a combination of UMN and LMN signs, with evidence of disease progression.30 Nerve conduction studies (NCS) and electromyography (EMG) are important clinical investigations, excluding potential mimic disorders,2 and identifying widespread LMN dysfunction, a cardinal feature of ALS. Specifically, LMN dysfunction may be heralded by the presence of ongoing activity (fibrillation potentials and positive sharp waves) and chronic neurogenic changes (large-amplitude, long-duration, polyphasic motor unit potentials), and if widespread appear to exhibit a high sensitivity and specificity for ALS.31 Importantly, the EMG changes may be evident sub-clinically, thereby enabling an earlier diagnosis of ALS.32 In addition, widespread fasciculations with a high firing frequency and increased frequency of double fasciculations, may also be a diagnostic feature of ALS,33 especially when combined with clinical features and disease progression.
Atypical MND phenotypes
Atypical MND phenotypes include progressive muscular atrophy, the clinically “pure” lower motor neuron phenotype, encompassing the flail-arm and some of the flail leg variants. The flail-limb variants are characterised by neurogenic weakness confined to the proximal upper limbs (flail-arm, at least for 24 months) or lower limbs (flail-leg, confined to lower limbs for at least 12 months).34,35 Importantly, one-third of PMA cases may develop UMN dysfunction, and while the overall prognosis for the flail-arm and leg variants is favourable,35 a progressive course akin to that evident in ALS may also be evident in PMA,36 underscoring the notion that PMA falls into the spectrum of MND diseases.
Of further relevance, primary lateral sclerosis refers to the pure UMN phenotype, characterised by a slowly progressive UMN syndrome (Table 1) affecting the spinal and bulbar regions with relative preservation of the lower motor neurons for at least four years after symptom onset.37,38 Importantly, lower motor neuron signs may develop within four years of symptom onset, and this group is then classified as upper motor neuron predominant-ALS.38 The PBP phenotype remains localised within the bulbar region for a prolonged period (>6 months) and is characterised by female predominance and UMN bulbar dysfunction, although clinical features of ALS may develop.39 The rates of survival for the UMN phenotypes of MND are typically prolonged, although significant functional impairment occurs.29
In conclusion, MND appears to be a clinically heterogeneous disorder with varied clinical presentation encompassing a range of upper and lower motor neuron dysfunction. The overlap in clinical features, along with evidence of disease progression underscores the notion that common pathophysiological processes underlie varied MND phenotypes. Discovering the processes that regulate the development of the varied clinical phenotypes, may yet result in development of adequate therapeutic strategies.
References
- Charcot J, Joffroy A. Deux cas d’atrophie musculaire progressive avec lesion de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arch Physiol Neurol Pathol 1869;2:744-54.
- Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet 2011;377:942-955.
- Renton Alan E, Majounie E, Waite A, et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011;72:257-68.
- DeJesus-Hernandez M, Mackenzie Ian R, Boeve Bradley F, et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011;72:245-56.
- Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 2011;7:639-49.
- Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002;59:1077-9.
- Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 1995;92:689-93.
- Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006;351:602-11.
- Turner MR, Hardiman O, Benatar M, et al. Controversies and priorities in amyotrophic lateral sclerosis. The Lancet Neurology 2013;12:310-22.
- Logroscino G, Traynor BJ, Hardiman O, et al. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol, Neurosurg, Psychiatry 2010;81:385-90.
- Ravits J, Paul P, Jorg C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007;68:1571-5.
- Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread Deconstructing motor neuron degeneration. Neurology 2009;73:805-11.
- Kuwabara S, Sonoo M, Komori T, et al. Dissociated small hand muscle atrophy in amyotrophic lateral sclerosis: frequency, extent, and specificity. Muscle Nerve 2008;37:426-30.
- Wilbourn AJ. The “split hand syndrome”. Muscle Nerve 2000;23:138.
- Li TM, Alberman E, Swash M. Clinical features and associations of 560 cases of motor neuron disease. J Neurol Neurosurg Psychiatry 1990;53:1043-5.
- de Carvalho M, Swash M. Cramps, muscle pain, and fasciculations: not always benign? Neurology 2004;63:721-3.
- Eisen A, Kim S, Pant B. Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve 1992;15:219-24.
- Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996-2000. J Neurol Neurosurg Psychiatry 2003;74:1258-61.
- Kurian KM, Forbes RB, Colville S, Swingler RJ. Cause of death and clinical grading criteria in a cohort of amyotrophic lateral sclerosis cases undergoing autopsy from the Scottish Motor Neurone Disease Register. J Neurol Neurosurg Psychiatry 2009;80:84-7.
- Scelsa SN, Yakubov B, Salzman SH. Dyspnea-fasciculation syndrome: early respiratory failure in ALS with minimal motor signs. Amyotroph Lateral Scler Other Motor Neuron Disord 2002;3:239-43.
- Czaplinski A, Strobel W, Gobbi C, Steck AJ, Fuhr P, Leppert D. Respiratory failure due to bilateral diaphragm palsy as an early manifestation of ALS. Medical Science Monitor 2003;9:CS34-6.
- Eisen A, Kuwabara S. The split hand syndrome in amyotrophic lateral sclerosis. J Neurol, Neurosurg Psychiatry 2012;83:399-403.
- Menon P, Kiernan MC, Vucic S. ALS pathophysiology: Insights form the split-hand phenomenon. Clin Neurophysiol 2014;125:186-93.
- Phukan J, Elamin M, Bede P, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry 2012;83:102-8.
- Snowden JS, Harris J, Richardson A, et al. Frontotemporal dementia with amyotrophic lateral sclerosis: a clinical comparison of patients with and without repeat expansions in C9orf72. Amyotroph Lateral Scler Frontotemporal Degener 2013;14:172-6.
- Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. The Lancet Neurology 2012;11:323-30.
- van Rheenen W, van Blitterswijk M, Huisman MH, et al. Hexanucleotide repeat expansions in C9ORF72 in the spectrum of motor neuron diseases. Neurology 2012;79:878-82.
- Al-Sarraj S, King A, Troakes C, et al. P62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathologica 2011;122:691-702.
- Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci 2014;37:433-42.
- Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2000;1:293-9.
- Costa J, Swash M, de Carvalho M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis:a systematic review. Arch Neurol 2012;69:1410-16.
- Krarup C. Lower motor neuron involvement examined by quantitative electromyography in amyotrophic lateral sclerosis. Clin Neurophysiol 2011;122:414-22.
- Mills KR. Characteristics of fasciculations in amyotrophic lateral sclerosis and the benign fasciculation syndrome. Brain 2010;133:3458-69.
- Vucic S, Kiernan MC. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2007;78:849-52.
- Wijesekera LC, Mathers S, Talman P, et al. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology 2009;72:1087-94.
- Visser J, van den Berg-Vos RM, Franssen H, et al. Disease course and prognostic factors of progressive muscular atrophy. Arch Neurol 2007;64:522-8.
- Pringle CE, Hudson AJ, Munoz DG, Kiernan JA, Brown WF, Ebers GC. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain 1992;115(Pt 2):495-520.
- Gordon PH, Cheng B, Katz IB, et al. The natural history of primary lateral sclerosis. Neurology 2006;66:647-53.
- Burrell JR, Vucic S, Kiernan MC. Isolated bulbar phenotype of amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis 2011;12:283-9.