



Key take home messages
- Research into the genetic and molecular mechanisms underlying MSA has grown significantly in recent years.
- No neurorestorative treatments for MSA are available to date but there are many ongoing trials targeting α-syn pathways.
- Currently the standard of care in this condition remains symptom control.
- Levodopa is widely used, but while a small number of patients have some response to it, its benefit is short lasting and can worsen MSA symptoms, including OH and dyskinesia. First line treatment of OH remains midodrine, but new drugs such as atomoxetine have shown greater improvement in OH compared to midodrine in randomised trials.
Multiple System Atrophy (MSA) is a neurodegenerative condition characterised by late onset, progressive, atypical Parkinsonism and/or cerebellar syndrome with autonomic failure. It has a prevalence of 4.4 in 100,000 individuals1 and affects males and females equally. Despite extensive research, the prognosis remains poor with average survival of eight years, although with careful management we are seeing patients live for 20 years after disease onset.2 Clinically, MSA usually presents either parkinsonian (MSA-P) or cerebellar ataxia (MSA-C) predominant features but in the later years combined clinical features are often found.
At present, management strategies are focused on symptom control with medication for parkinsonism, autonomic liability, bladder and bowel dysfunction and mood problems. The role of specialised MSA clinics and working closely with the MSA Trust nurses is most important in symptom management as discussed in another article in ACNR here. Disease modifying treatments that can stop or reverse the disease have yet to be identified. In order to develop new neurorestorative drugs and attempt to halt or reverse the pathology we need to advance our understanding of the mechanisms underlying MSA. Cell and animal models, and genetic studies have provided interesting results, yet no treatments have been demonstrated to slow or reverse the disease in humans. This article provides an update into recent drug trials and potential therapeutic targets linked to advances in the molecular and genetic aspects of MSA.
MSA pathways and therapeutic targets
The neuropathological characterisation of MSA has led to significant advances in research revealing the accumulation of abnormally misfolded α-synuclein (α-syn) and the pathognomonic formation of glial cytoplasmic inclusions (GCIs).3 Native α-syn is a soluble protein present in normal brain tissue, usually in neurons, and believed to be involved in the pre-synapse and neurotransmission via the SNARE complex.4,5 Although the conformational states of α-syn in different cell compartments are not well known, the existence of a balanced combination of monomers and tetramers resistant to aggregation has been proposed.6 The mechanisms by which α-syn, a neuronal protein, ends up in the oligodendroglia and propagates from cell-to-cell and between cell types remains unclear. How this leads to neuronal degeneration is key to finding therapeutic targets in MSA. There is growing evidence that oligodendroglial pathology is the primary event in MSA leading to neurodegeneration and is involved in multiple pathways such as microtubule stabilisation and myelination.7 In MSA patients, P25α accumulates early in the oligodendrocytes’ body, rather than in the myelin where it would normally be located. It has been shown to promote oligomerisation and aggregation of α-syn.8,9
Another protein found to be involved early in α-syn aggregation is β-III tubulin. β-III tubulin is identified in GCIs in MSA patients and furthermore, α-syn binds directly to β-III tubulin via an α-syn binding site forming insoluble α-syn aggregates. Mouse model and mouse derived cell cultures show evidence that α-syn binding to β-III tubulin could be the initial event leading to α-syn self-aggregation.10 Inhibition of this binding site has been shown to prevent α-syn accumulation.11 These results suggest that β-III tubulin is an important candidate for therapeutic targets with the potential to halt neurodegenerative changes in MSA.
The α-syn oligomerisation ultimately leads to oligodendrocytes’ apoptosis. The apoptosis itself is mediated by several proteins such as sirtuin 2 (SIRT2), a tubulin deacetylase exclusively found in oligodendroglia with a promoting role in neurodegeneration.12 Ultimately, targeting the proteins involved in apoptosis could prove a good therapeutic strategy. It appears that disease-modifying treatments should target one or a combination of the following: α-syn aggregation and propagation, neuronal demyelination, inflammation, apoptosis and eventually halt neurodegeneration.
Genetic targets in MSA
Although a few familial cases with Mendelian inheritance have been reported13,14 MSA is usually a sporadic disorder. Extensive work has been undertaken studying the genetics of MSA in recent years. As abnormal α-syn deposited as GCI’s is the hallmark of MSA, several studies have looked at SNCA mutations (gene encoding for α-syn) as a cause of disease. Despite SNCA mutations (A30P, H50Q, G51D, A53T, A53E,) being associated with familial Parkinson’s disease (PD) leading to α-syn aggregation and cell toxicity, no SNCA mutations have been identified in true sporadic MSA. However, several families with SNCA mutations (e.g. G51D, SNCA triplications) presented with clinical and/or neuropathological similarities of autosomal dominant PD and MSA,15,16,17 suggesting a possible link between the two conditions. A subsequent study targeting multiplications in SNCA in pathologically confirmed MSA cases did not identify any SNCA duplication or triplication.18 Nevertheless, a study looking at single nucleotide polymorphism (SNP) identified SNPs in SNCA associated with increased risk of MSA in the Caucasian population19 and replicated in other European studies,20 but not replicated in Chinese or Korean populations.21 Screening for other PD causal genes (MAPT, LRRK2, PINK1) has not yet revealed any association with MSA.22-24
Other genes believed to increase predisposition to MSA included COQ2 mutations. A study combining linkage analysis and whole-genome sequencing found the mutations M128V-V393A/M128V-V393A (homozygous) and R337X/ V393A (compound heterozygous state) in two Japanese MSA families.25 Also, the V393A variant was found over-represented in a cohort of Japanese MSA patients, suggesting it as a risk factor for MSA. However, several other studies were not able to replicate these results.26,27
Several recent reports have focused on prion-like mechanisms in synucleinopathies, but no variants in the prion protein gene (PRNP) were found to be associated with increased risk of MSA.28 However no large scale study has been carried out looking at the PRNP gene in MSA. Finally, other genes have been screened for their association with MSA including several Spinocerebellar Ataxia (SCA) genes. There is no strong evidence to suggest an association with SCA and MSA at present, but larger studies in pathologically confirmed MSA cases are needed.
Therapies targeting α-synuclein
Several therapeutic strategies targeting α-syn aggregation and propagation have been attempted, but most were equivocal or failed. The only trial to date that reduced disease progression (based on UMSARS score) was a small-randomised mesenchymal stem cell (MSC) treatment. This suggests that MSC could be a potential treatment in MSA but there are limitations to the study due to the small number of patients involved,29 the associated ischaemic side effects with the drug delivery and uncertainty over how the MSC targets α-syn.
In transgenic MSA mouse α-syn aggregation was significantly reduced after administration of rifampicin.30 However the trials with rifampicin in humans did not show any significant benefit. One hundred patients with possible or probable MSA were randomly assigned to either rifampicine (50 patients) or placebo (50 patients). The primary outcome measured was a change in the UMSARS score. This study showed that rifampicin did not slow progression of MSA.31
Lithium showed promising results in α-syn aggregation in mouse models by inducing autophagy and α-syn clearance. However, in a placebo-controlled double blind trial it did not pass the safety and tolerability in MSA and further lithium studies for this condition are not encouraged.32 Selective serotonin reuptake inhibitors (SSRI) have been shown to prevent α-syn aggregation and propagation to oligodendroglia. Sertraline, Paroxetine and Fluoxetine are the most studied examples. In in-vivo transgenic mice models, fluoxetine reduced α-syn aggregation, gliosis and demyelination together with an increase in glial and brain derived neurotrophic factors. Clinically, the mouse showed improved motor and behavioural deficits.33 Sertraline was shown to inhibit the propagation of α-syn to oligodendroglia in cell culture models.34 Paroxetine is the only SSRI tested in MSA patients. A double-blind placebo-controlled randomised trial showed that the paroxetine treated group had improved motor and speech symptoms compared to placebo.35 A new study currently at recruitment stage will assess Fluoxetine in MSA patients. All these studies show that SSRIs could represent promising therapeutic targets in MSA.
Interesting results were obtained using non-steroidal anti-inflammatory drugs (NSAIDs) to inhibit the formation of α-syn fibrils and even destabilisation of preformed α-syn fibrils in a dose-dependent manner.36 NSAIDs have been extensively used in other conditions, have a well-known side effects profile and are inexpensive. All these aspects make them a potential therapeutic option and further studies are needed. As microglia activation plays an important role in MSA, a trial assessing the role of minocycline was conducted. Minocycline did not show any improvement in patients’ motor symptoms or UMSARS. However, the group treated with minocycline showed a reduction in [11C](R)-PK11195 PET activity suggesting that minocycline has an effect on microglial activation and requiring further research.37
Another study used nocodazole (microtubule depolymerisation drug) to stop α-syn accumulation on the basis that β-III tubulin was shown to be involved early in the disease by binding to α-syn forming insoluble complexes leading to MSA pathology. It was shown that in cultured cells nocodazole prevented the accumulation of insoluble α-syn when the drug was given before the formation of α-syn aggregates, but had no effect when given at later stages.10 No P25α targeting treatment has been tried yet.
On the premise that SIRT2 is involved in oligodendroglial apoptosis and promotes neurodegeneration one study assessed the role of a SIRT2 inhibitor. They showed that the SIRT2 inhibitor partially prevented cellular apoptosis in a cell culture model, but the exact mechanism is still unclear.12
Recently, it has been shown that neurosin (human kalikrein6 -KLK6), a serine protease present in many human tissues including the astrocytes, is able to cleave α-syn aggregates. Furthermore, the down-regulation of neurosin leads to accumulation of α-syn. In the MSA mouse brain model the genetically stabilised form of neurosin allowed it to be delivered systemically and led to the reduction of α-syn aggregation and propagation, together with improvement in clinical features such as demyelination and behaviour.38
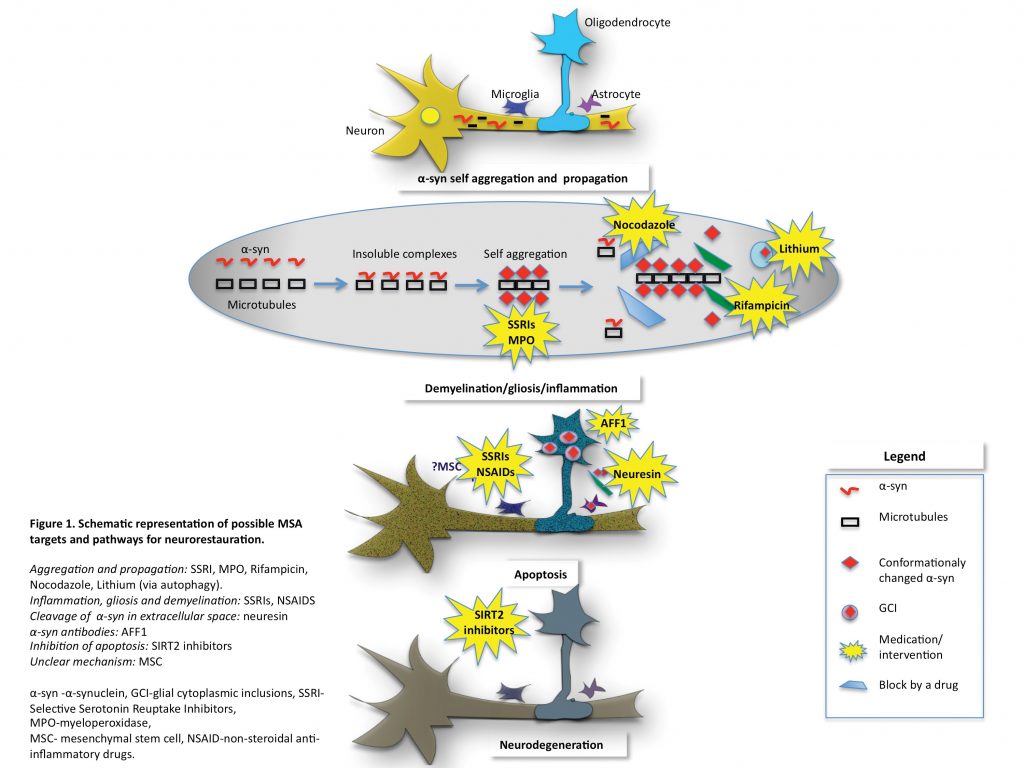
Several studies have or are looking at active vaccination in an attempt to stop α-syn aggregation and propagation. Mandler et al used short peptides AFF1 in a MSA transgenic mouse model and showed that active vaccination with AFF1 resulted in the production of anti- α-syn antibodies capable of identifying α-syn in oligodendroglia. In their animal model, this process led to a reduction of α-syn accumulation, demyelination and neurodegeneration.39 A new European project (The SYMPATH project) is currently assessing a vaccine targeting α-syn (AFFITOPE) in PD and MSA in humans.
Therapies targeting symptom control in MSA
Symptomatic treatment remains the current standard of care in MSA. Trials targeting improvement in symptom control published in the last two years are summarised in Table 1. The symptom control efforts are focused on dopamine replacement in the parkinsonian type MSA, management of the autonomic dysfunction, bladder and bowel care, sleep, breathing and mood problems.
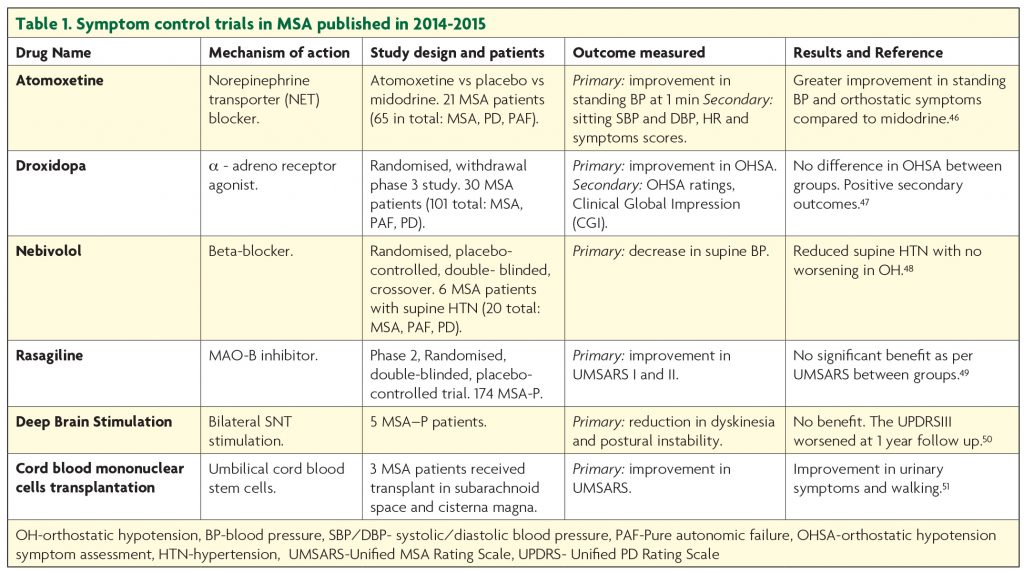
Part of the diagnostic criteria for MSA is the poor response to levodopa. However, in a recent USA study, about half of the patients, particularly with probable MSA-P type reported benefit from Levodopa.40 The European MSA Study Group demonstrated a three years sustained Levodopa response in 31% of patients.2 A retrospective review of pathologically confirmed MSA cases showed benefit from Levodopa in about 30%, though this was not long lasting.41 Levodopa is known to cause fewer hallucinations in MSA than PD but it can worsen orthostatic hypotension42 and induce dyskinesia.43 So far, there has been no randomised controlled trial assessing the efficacy of levodopa or dopamine agonists in MSA but it appears that levodopa provides short lasting, modest benefit, in a small number of patients.
Orthostatic hypotension (OH) represents one of the major diagnostic criteria for MSA and is a very disabling symptom for patients. So far, midrodrine (α1-adrenoceptor agonist) is the only medication that has shown improvement in neurogenic OH in randomised, double blinded placebo studies at doses up to 30 mg per day.44 Patients on midodrine should have regular blood pressure monitoring as it can induce supine hypertension. Also, non-pharmacological measures, such as higher salt and fluid intake, smaller but more frequent meals together with head raising to 30° in bed, wearing elastic stockings and a special set of exercises, improve symptoms and should be recommended.
Symptomatic interventions tackling neurogenic bladder include intermittent self-catheterisation if post-void residual volume is >100 ml. Anticholinergics and α-adrenergic antagonists are indicated if post voidal residue is <100 ml. Side effects include urinary retention and worsening OH. In severe cases, Botox injections can be administered to the detrusor muscle or urethral sphincter. Botox injections are also indicated for the management of dystonia, camptocormia and excessive salivation.
For patients experiencing breathing problems continuous positive air pressure (CPAP) is beneficial or tracheostomy in advanced stages. First line treatment for REM sleep disorders is clonazepam45 but other medication such as zopiclone, melatonin and temazepam can be used as second line. Selective Serotonin Reuptake Inhibitors (SSRIs) are recommended for the management of depression together with psychological support for the patient and the family. Levodopa can also have a beneficial effect on depression.
References
- Schrag A, Ben-Shlomo Y, and Quinn NP, Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet, 1999;354(9192):1771-5.
- Wenning GK, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol, 2013;12(3):264-74.
- Wenning GK, et al. Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol, 2008;64(3):239-46.
- Burre J, et al. Properties of native brain alpha-synuclein. Nature, 2013;498(7453):E4-6; discussion E6-7.
- Lee FJ, et al. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J, 2001;15(6): 916-26.
- Bartels T, Choi JG, and Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature, 2011;477(7362):107-10.
- Ovadi J. Orosz F. An unstructured protein with destructive potential: TPPP/p25 in neurodegeneration. Bioessays, 2009;31(6):676-86.
- Hasegawa T, et al. Role of TPPP/p25 on alpha-synuclein-mediated oligodendroglial degeneration and the protective effect of SIRT2 inhibition in a cellular model of multiple system atrophy. Neurochem Int, 2010;57(8):857-66.
- Wang Y, et al. Phosphorylated alpha-synuclein in Parkinson’s disease. Sci Transl Med, 2012;4(121):121ra20.
- Nakayama K, Suzuki Y, and Yazawa I, Binding of neuronal alpha-synuclein to beta-III tubulin and accumulation in a model of multiple system atrophy. Biochem Biophys Res Commun, 2012;417(4):1170-5.
- Suzuki Y, et al. beta-III Tubulin fragments inhibit alpha-synuclein accumulation in models of multiple system atrophy. J Biol Chem, 2014;289(35):24374-82.
- Harting K and Knoll B. SIRT2-mediated protein deacetylation: An emerging key regulator in brain physiology and pathology. Eur J Cell Biol, 2010;89(2-3):262-9.
- Hara K, et al. Multiplex families with multiple system atrophy. Arch Neurol, 2007;64(4):545-51.
- Wullner U, et al. Probable multiple system atrophy in a German family. J Neurol Neurosurg Psychiatry, 2004;75(6):924-5.
- Kiely AP, et al. alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol, 2013;125(5):753-69.
- Kiely AP, et al. Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener, 2015;10:41.
- Pasanen P, et al. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging, 2014;35(9):2180 e1-5.
- Lincoln SJ, et al. Quantitative PCR-based screening of alpha-synuclein multiplication in multiple system atrophy. Parkinsonism Relat Disord, 2007;13(6):340-2.
- Scholz SW, et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol, 2009;65(5):610-4.
- Ross OA, et al. Reply to: SNCA variants are associated with increased risk of multiple system atrophy. Ann Neurol, 2010;67(3):414-5.
- Sun Z, et al. SNP rs11931074 of the SNCA gene may not be associated with multiple system atrophy in Chinese population. Int J Neurosci, 2015;125(8):612-5.
- Ozelius LJ, et al. G2019S mutation in the leucine-rich repeat kinase 2 gene is not associated with multiple system atrophy. Mov Disord, 2007;22(4):546-9.
- Morris HR, et al. Multiple system atrophy/progressive supranuclear palsy: alpha-Synuclein, synphilin, tau, and APOE. Neurology, 2000;55(12):1918-20.
- Brooks JA, et al. Mutational analysis of parkin and PINK1 in multiple system atrophy. Neurobiol Aging, 2011;32(3):548.e5-7.
- Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med, 2013;369(3):233-44.
- Schottlaender LV and Houlden H. Mutant COQ2 in multiple-system atrophy. N Engl J Med, 2014;371(1):81.
- Sharma M, Wenning G, and Kruger R. Mutant COQ2 in multiple-system atrophy. N Engl J Med, 2014;371(1):80-1.
- Shibao C, et al. PRNP M129V homozygosity in multiple system atrophy vs. Parkinson’s disease. Clin Auton Res, 2008;18(1):13-9.
- Konno M, et al. Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol Neurodegener, 2012;7:38.
- Ubhi K, et al. Rifampicin reduces alpha-synuclein in a transgenic mouse model of multiple system atrophy. Neuroreport, 2008;19(13):1271-6.
- Low PA, et al. Efficacy and safety of rifampicin for multiple system atrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol, 2014;13(3):268-75.
- Sacca F, et al. A randomized clinical trial of lithium in multiple system atrophy. J Neurol, 2013;260(2):458-61.
- Ubhi K, et al. Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of alpha-synucleinopathy. Exp Neurol, 2012;234(2):405-16.
- Konno M, et al. Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol Neurodegener, 2012;7:38.
- Friess E, et al. Paroxetine treatment improves motor symptoms in patients with multiple system atrophy. Parkinsonism Relat Disord, 2006;12(7):432-7.
- Hirohata M, et al. Non-steroidal anti-inflammatory drugs have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. Neuropharmacology, 2008;54(3):620-7.
- Dodel R, et al. Minocycline 1-year therapy in multiple-system-atrophy: effect on clinical symptoms and [(11)C] (R)-PK11195 PET (MEMSA-trial). Mov Disord, 2010;25(1):97-107.
- Spencer B, et al. A brain-targeted, modified neurosin (kallikrein-6) reduces alpha-synuclein accumulation in a mouse model of multiple system atrophy. Mol Neurodegener, 2015;10(1):48.
- Mandler M, et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener, 2015;10(1):10.
- Low PA, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol, 2015;14(7):710-9.
- Wenning GK, et al. Clinicopathological study of 35 cases of multiple system atrophy. J Neurol Neurosurg Psychiatry, 1995;58(2):160-6.
- Wenning GK, et al. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain, 1994;117(Pt 4):835-45.
- Boesch SM, et al. Dystonia in multiple system atrophy. J Neurol Neurosurg Psychiatry, 2002;72(3):300-3.
- Low PA, et al. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. Jama, 1997;277(13):1046-51.
- Ghorayeb I, Bioulac B, and Tison F. Sleep disorders in multiple system atrophy. J Neural Transm (Vienna), 2005;112(12):1669-75.
- Ramirez CE, et al. Efficacy of atomoxetine versus midodrine for the treatment of orthostatic hypotension in autonomic failure. Hypertension, 2014;64(6):1235-40.
- Biaggioni I, et al. Randomized withdrawal study of patients with symptomatic neurogenic orthostatic hypotension responsive to droxidopa. Hypertension, 2015;65(1):101-7.
- Okamoto LE, et al. Nebivolol, but not metoprolol, lowers blood pressure in nitric oxide-sensitive human hypertension. Hypertension, 2014;64(6):1241-7.
- Poewe W, et al. Efficacy of rasagiline in patients with the parkinsonian variant of multiple system atrophy: a randomised, placebo-controlled trial. Lancet Neurol, 2015;14(2):145-52.
- Zhu XY, et al. Effects of deep brain stimulation in relatively young-onset multiple system atrophy Parkinsonism. J Neurol Sci, 2014;342(1-2):42-4.
- Wu SH, et al. Preliminary results of cord blood mononuclear cell therapy for multiple system atrophy: a report of three cases. Med Princ Pract, 2014;23(3):282-5.