


Abstract
Epstein Barr virus (EBV) and Varicella Zoster virus (VZV) co-infection of the central nervous system has not previously been reported. We describe a patient who presented with EBV meningitis complicated by bilateral optic neuritis and facial palsy who developed reactivation of VZV without skin manifestations. The VZV infection caused cerebral vasculitis with multiple cerebral infarcts and longitudinally extensive transverse myelitis leading to paraparesis. We postulate that VZV reactivation should be considered as a possible cause of neurological deterioration in patients with acute EBV infection so that prompt antiviral treatment can be initiated.
Case report
A 20-year-old previously well woman with a history of uncomplicated chicken pox as a child presented to another hospital’s emergency department with a two-week history of sore throat and fevers not responding to oral amoxicillin and clavulanic acid. She was febrile, with enlarged tonsils and cervical lymphadenopathy. Epstein Barr virus (EBV) IgM was positive confirming a diagnosis of infectious mononucleosis. Throat culture was negative. She was discharged on day three on oral penicillin after brief treatment with intravenous antibiotics.
On day six, she represented with fevers and headache. She had meningism and photophobia but the remainder of the neurological examination was normal. There was no skin rash. A lumbar puncture (LP) yielded turbid cerebrospinal fluid (CSF) with elevated protein and mononuclear cell count (Table 1). She was commenced on intravenous ceftriaxone and penicillin. After seven days, CSF culture was negative and antibiotics were ceased.
On day 13, her visual acuity deteriorated to count fingers in the right eye and hand movements in the left eye. She had mydriasis of both pupils with a relative afferent pupillary defect in the left eye. There was a complete right sided lower motor neurone facial droop. The remainder of her examination was normal. Repeat LP showed a greater lymphocytosis with further elevation of CSF protein (Table 1). The patient was transferred to our hospital.
The following day she developed acute urinary retention. Resting tachycardia was noted at 130 bpm with a postural blood pressure drop of 120/78 mmHg to 75/40 mmHg despite normal hydration status. Pyridostigmine was commenced up to 120mg tds with improvement in postural blood pressure. Hyponatraemia developed reaching a nadir of 116mmol/L with serum osmolality 257 mmol/kg, urinary sodium 79 mmol/L, urine osmolality 489 mmol/kg suggesting syndrome of inappropriate antidiuretic hormone (SIADH). This resolved with fluid restriction.
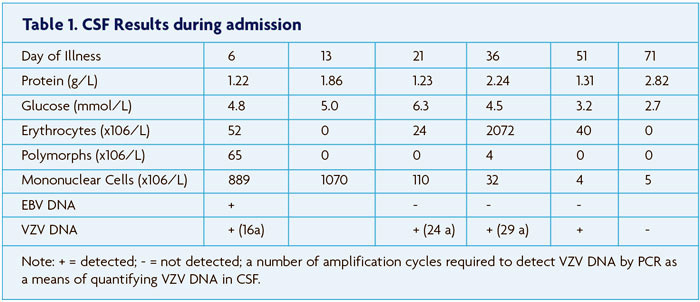
MRI brain with gadolinium (Figure 1A) demonstrated intense bilateral optic nerve sheath enhancement with lesser basal meningeal and internal auditory canal enhancement. She was treated with intravenous methylprednisolone 1g/day for three days followed by oral dexamethasone (2gms qid and weaned over several days). Extensive testing for other inflammatory and infective causes of optic neuritis was negative. EBV polymerase chain reaction (PCR) of the initial CSF was positive, as was VZV PCR.
One week later, by day 21, there was minor improvement in the facial weakness and visual acuity, and bladder function returned. Repeat CSF examination showed a decrease in protein and cell count (Table 1). Repeat MRI revealed reduction in optic nerve enhancement. An acute infarct in the anterior aspect of the right thalamus was seen on diffusion weighted imaging.
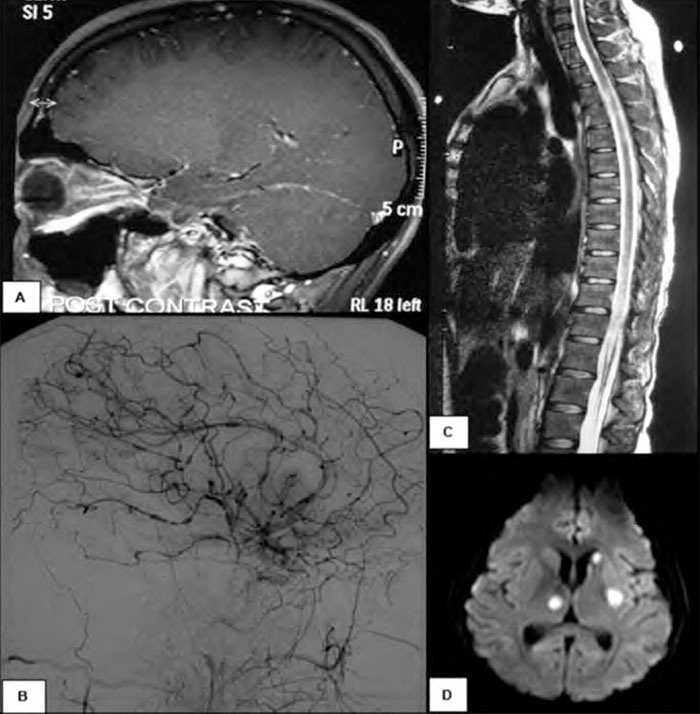
On day 36 she became febrile to 38.7 degrees and had mild left lower limb weakness. Repeat CSF examination showed a further increase in protein, and reduction in cell count (Table 1).VZV PCR was positive but EBV PCR was negative. MRI demonstrated several subcentimetre acute infarcts within the right cerebellar hemisphere and the left external capsule. There was further meningeal enhancement with regions of nodularity, but optic nerve sheath enhancement had resolved. A formal cerebral angiogram (Figure 1B) revealed extensive beading and variation in calibre of the cerebral arteries particularly supratentorially, sparing the external carotid branches, indicating severe vasculitis. She was commenced on intravenous acyclovir 10mg/kg tds, intravenous methylprednisolone 1g daily for three days and intravenous immunoglobulin 20g daily for five days.
The next day, on day 37, she had a complete paraplegia with a complete motor and sensory level at T6 and there was a new right 6th nerve palsy. MRI spinal cord (Figure 1C) showed longitudinally extensive T2 signal within the entire thoracic cord with pachymeningeal enhancement but no enhancement of the cord. The first of five plasma exchange treat- ments was started and intravenous cyclophosphamide 1g was given.
On day 38, a bulbar palsy was noted with dysarthria, dysphagia, decreased palatal movement and decreased gag reflex. This improved over the following week with no change to treatment. On day 45, treatment was changed to prednisolone 30mg/day and oral valacyclovir 1g tds.
MRI on day 58 revealed several new foci of abnormal signal on diffusion-weighted imaging within the left cerebellar hemisphere, the right precentral gyrus, the right thalamus and left caudate lobe. A SPECT scan was consistent with active cerebritis.
By day 71, the patient was able to read large print with her right eye. Her paraparesis had not improved. Repeat MRI spine was unchanged. She now developed sudden onset right arm weakness and expressive aphasia. MRI showed new acute infarcts in the left basal ganglia, right thalamus and left head of caudate nucleus (Figure 1D). Repeat CSF (Table 1) showed a further elevation of protein. VZV DNA was not detected. Flow cytometry of the CSF was normal. A brain biopsy of the right frontal lobe was performed which revealed no evidence of vasculitis, lymphocytic infiltrate or viral inclusions although there was some evidence of reactive T cells. EBV and VZV staining was negative.
After discussion with the patient and her family, treatment with antiviral agents and immunomodulatory therapy was discontinued and the patient was discharged home. At the time of discharge the neurological examination showed a dilated left pupil with a pale optic disc. Visual acuity was count fingers in the left eye and perception of light in the right eye. She had aright facial palsy, right arm weakness, expressive dysphasia and T6 paraparesis. At follow-up twelve months later there had been no further neurological events and although she had marked improvement in her right arm weakness and aphasia there had been no improvement in her visual acuity or paraparesis.
Discussion
The patient presented with typical features of infectious mononucleosis confirmed by positive EBV IgM antibodies. EBV infection may be complicated by meningitis, encephalitis, myelitis, optic neuritis and cranial nerve palsies, most commonly the facial nerve.1 It can also be complicated by autonomic neuropathy and salt-wasting nephropathy.2 Our patient had meningitis, bilateral optic neuritis, facial nerve palsy autonomic dysfunction and hyponatraemia. EBV infection of the CNS was confirmed with positive CSF PCR. Randomised controlled studies demonstrating efficacy of anti-viral treatment or corticosteroids in EBV infection of the CNS are lacking.3,4 Corticosteroids in many case reports have good clinical effect. Treatment with corticosteroids in our patient initially resulted in clinical improvement of the optic neuritis, meningitis and facial palsy, a decrease in inflammation in the CSF and decreased enhancement on the MRI.
VZV PCR of the CSF was also positive initially, later becoming negative. VZV serology showed positive IgG and equivocal IgM confirming prior infection. There was no evidence of a rash during the current illness. Co-infection with VZV and EBV has been previously reported in a small number of immuno-competent and immunocompromised patients.5 In these patients, the inflammatory response to the VZV was thought to trigger EBV reactivation. In our patient there was serolog- ical and clinical evidence of primary EBV infection and past VZV infection, thus the inflammatory response to EBV may have triggered reactivation of latent VZV.
Isolated cerebral vasculitis has not been reported with EBV infection. Systemic vasculitis with EBV infection may occur but usually in the context of immunosuppression. VZV is more frequently causative of cerebral vasculitis and mixed large and small artery involvement is usual.6 Multifocal vasculopathy mostly occurs in immunocompromised individuals although there have been several case reports of multifocal cerebral infarcts in immunocompetent patients with VZV vasculitis without skin involvement.7,8 Consanguinity was present in the family, raising the possibility of a recessive immunodeficiency disorder. Immunoglobulins tested during the illness were normal. Lymphocyte subsets showed marked reduction in CD4 Helper cells and some reduction in pan CD3 positive T cells.
Treatment of VZV vasculitis is aimed at suppressing viral replication with acyclovir. Optimal treatment is not currently defined but in a recent case series of 23 patients, 66% improved or stabilised with acyclovir alone compared with 75% treated with acyclovir in combination with corticosteroids.6 Due to the severity of the vasculitis in our patient, treatment with IVIg was added as was cyclophosphamide. In retrospect, the cyclophosphamide may have paradoxically exacerbated the vasculitis by allowing VZV to proliferate unchecked. The vasculitis appears to have remitted with cessation of both immunosuppressant medications and acyclovir. The amount of VZV DNA in the CSF (Table 1) was declining prior to treatment with acyclovir being instigated. VZV viral load in the CSF as detected by PCR has been correlated with the severity of neurologic disease.9
Myelopathy in our patient may be due to either EBV or VZV. Transverse myelitis is the least common neurological complication of EBV but occurs more frequently in younger individuals than adults.1 Myelitis in VZV infection has been found to be due to parenchymal invasion by the virus and is more severe in immunocompromised individuals.10 It may also be secondary to vasculitis.
Conclusion
In summary, we present a previously healthy young woman who presented with acute infectious mononucleosis complicated by meningitis, optic neuritis, facial nerve palsy and hyponatraemia. Reactivation of VZV caused cerebral vasculitis with extensive cerebral infarction, myelitis and poor neurologic outcome. This combination of complicated EBV infection with reactivation of VZV has not previously been reported. VZV infection should be considered as the cause of neurologic illness following EBV infection even in patients without a rash. This case questions whether antiviral treatment alone or in combination with cyclophosphamide should be used to treat the cerebral vasculitis which can complicate VZV CNS infection.
References
- Connelly K, DeWitt L. Neurologic complications of infectious mononucleosis. Pediatr Neurol. 1994;10:181-4.
- Corssmit EP, Leverstein-van Hall MA, Portegies P, Bakker P. Severe neurological complications in assocation with Epstein-Barr virus infection. J Neurovirol. 1997;3:460-4.
- Portegies P, Corssmit N. Epstein-Barr virus and the nervous system. Curr Opin Neurol. 2000;13:301e4.
- Volpi A. Epstein-Barr virus and human herpesvirus type 8 infections of the central nervous system. Herpes. 2004;11(Suppl. 2):120Ae7.
- Weinberg A, Bloch KC, Li S, Tang y, Palmer M, Tyler KL. Dual infections of the central nervous system with Epstein-Barr virus. J Infect Dis. 2005;191:234-7.
- Nagel MA, Cohrs RJ, Mahalingam R et al. The varicella zoster virus vasculopathies. Neurology. 2008;70:853-860.
- Hattori H, Higuchi Y, Tsuji M. Recurrent strokes after varicella. Ann Neurol. 2000; 47:136.
- Gilden DH, Kleinschmidt-DeMasters BK, Wellish M, Hedley-Whyte ET, Rentier B, Mahalingam R. Varicella zoster virus, a cause of waxing and waning vasculitis: the N Engl J Med case 5-1995 revisited. Neurology. 1996;47:1441-6.
- Aberle SW, Aberle JH, Steininger C, Puckhammer-Stöckl E. Quantitative real time PCR detection of Varicella-zoster virus DNA in cerebrospinal fluid in patients with neuro- logical disease. Med Microbiol Immunol. 2005;194:7-12.
- Gilden DH, Kleinschmidt-DeMasters BK, LaGuardia JJ, Mahalingam R, Cohrs RJ. Neurological Complications of the reactivation of varicella-zoster virus. New Engl J Med. 2000;342:632-45.