

Summary
- ODDD is a complex genetic disorder which illustrates the effects of a single mutation on multiple tissues: a multidisciplinary approach to management is necessary.
- The associated radiological changes on magnetic resonance images (MRI) of the brain are distinctive but may easily be misinterpreted or go unrecognised.
- Early recognition of ODDD allows the prevention and treatment of clinical manifestations and complications.
Oculo-dento-digital dysplasia (ODDD), also known as oculodentoosseous dysplasia (ODOD), is a rare genetic disorder affecting multiple tissues. It is characterised by multiple, variable craniofacial, limb, ocular and dental anomalies which are often associated with neurological disorder. Some features are evident at birth while others become evident with age.
The disorder results from mutations in the GJA1 gene, located on chromosome 6, which encodes for the gap junction protein, connexin43 (Cx43). It is inherited as an autosomal dominant trait in the majority of cases. Less commonly, sporadic forms occur which may be related to advanced paternal age. Autosomal recessive inheritance has been reported rarely, but this remains to be confirmed. There are more than 60 known mutations, mostly missense in type. Fewer than 1000 patients with ODDD have been reported in the literature, and the prevalence of the disorder is uncertain.
The story
A 50-year-old right-handed woman, who works part-time in a bakery, was referred because of progressive disturbance of gait of six years duration. This had started as a limp on the left leg, noticed after recovering from septic shock associated with bursitis of the left knee. Her gait disturbance was initially attributed to this dramatic episode, but further assessment revealed a potentially relevant background history.
She was born with bilateral partial syndactyly of the toes and fingers and complete syndactyly of the 4th and 5th fingers of the left hand. This was later surgically corrected. The proximal phalanx of the little finger of the right hand was absent (Figure 1). Her little fingers and toes remained very small. At the time of birth, her mother and father were aged 31 and 30 years respectively. Neither her parents nor her older brother have any developmental anomalies.
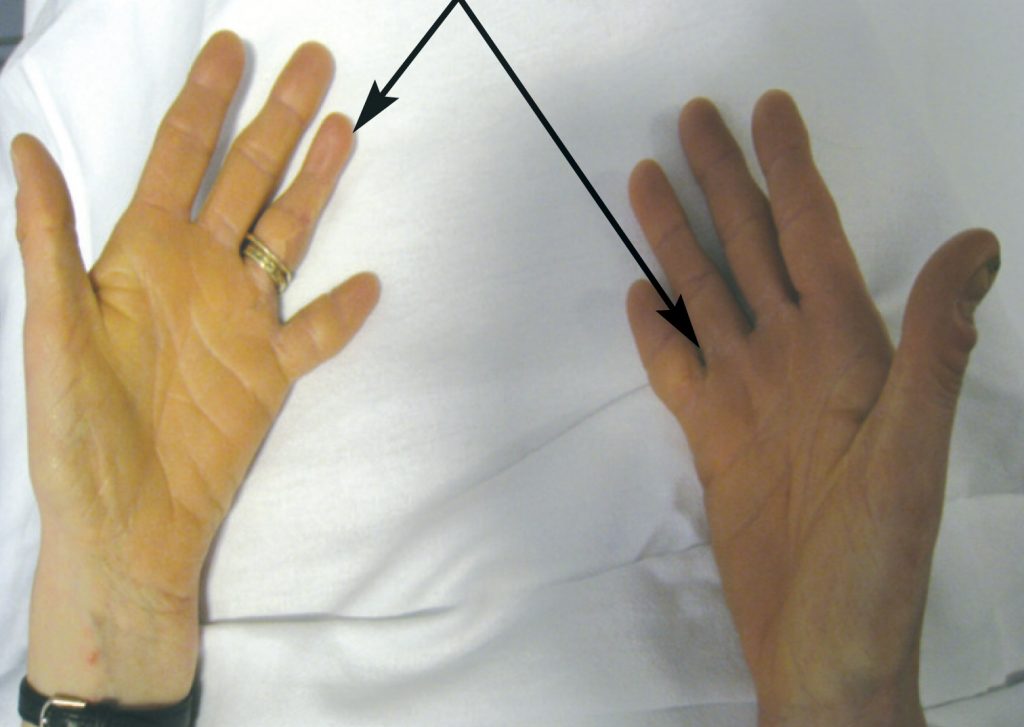
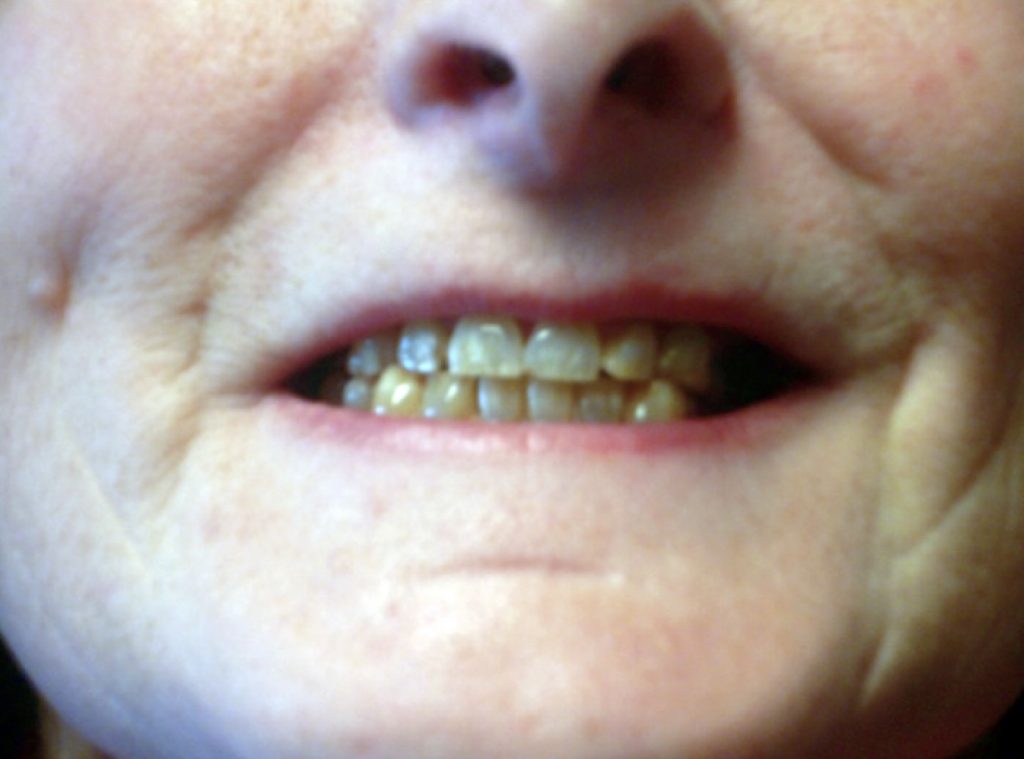
As a child, she developed a squint which was surgically corrected. Between the ages of 14-16, she suffered from epilepsy treated with Phenobarbital. The enamel of her teeth was deficient and she had lost four permanent teeth by the age of 24. She was prone to frequent diarrhoea, urinary frequency, nocturia, urgency, and precipitancy with occasional urinary incontinence. She had primary infertility, due to malformation of the Fallopian tubes. She was never sporty.
Examination revealed microphthalmia, a low bridge of nose with low insertion of columella (Figures 2, 3) and small teeth, in addition to the sketetal abnormalities described above. She had thickened skin over the palms and soles. She had a mild convergent squint, manifest on the left and latent on the right. There was spasticity of all four limbs with mild bilateral weakness of finger abduction, hip flexion, and knee flexion, very brisk tendon reflexes with a few beats of clonus at the ankles, and bilaterally extensor plantar responses. Coordination and superficial and deep sensations were intact. Her gait was spastic (stiff and hyperextended).

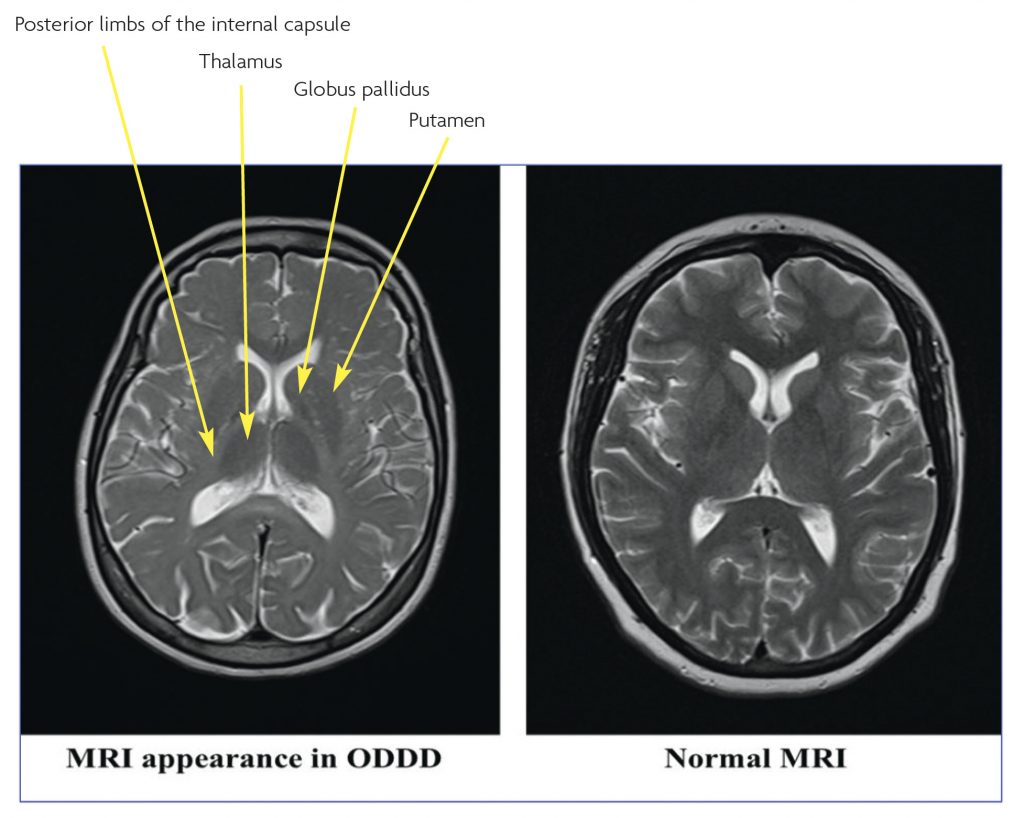
Brain MRI was initially reported to be normal, but on specialist review (initially at our neuroradiology meeting) there were subtle T2 hyperintensities in the subcortical white matter and T2 hypointensity of the basal ganglia (Figure 4). MRI images of the spine were normal.
The diagnosis was suspected clinically. A sequencing analysis of the GJA1 gene undertaken at John Hopkins DNA diagnostic laboratory in the United States, revealed a heterozygous mutation, c.460 A>G (Thr154Ala) in the GJA1 gene (6q22-q23), confirming the diagnosis of oculo-dento-digital dysplasia.
Discussion
The patient suffers from the sporadic form of ODDD. The disorder affects several tissues, including the eyes, nose, teeth, fingers, toes, the skin of the palms and soles, fallopian tubes and nervous system. The Fallopian anomaly has not been described in ODDD previously.
The most common anomalies in ODDD are ophthalmic, nasal, dental and digital. Seventy eight percent of affected families display features in more than two of these categories.1 The characteristic facial appearance is evident in 92% of the families.1 Eye findings are present in 68% and include short palpebral fissure (the anatomic name for the separation between the upper and lower eyelids), epicanthal folds (skin of the upper eyelid covering the inner corner of the eye), hyper- or hypotelorism (increased or decreased distance between the eyes), microphthalmia with microcornea and iris abnormalities. Gaze palsies and squint may also occur. Some patients develop cataracts, glaucoma and blindness secondary to glaucoma.1,2 Nasal features are also prominent in ODDD and may include, a thin nose, usually with hypoplastic alae nasi (the expanded outer wall of cartilage on each side of the nose), small anteverted nares (upturned nostrils), and prominent columella nasi (the fleshy lower margin of the nasal septum). Abnormal primary and permanent dentition with microdontia, partial anodontia, enamel hypoplasia, multiple caries and early tooth loss are evident in 70% of the affected families.1 Digital malformations occur very commonly and are seen in 80% of the affected families.1 Bilateral complete syndactyly of the fourth and fifth fingers (type III syndactyly) is characteristic. The third finger may also be involved. Syndactyly of the third and fourth toes may be present. Hypoplasia of the middle or distal phalanges or aplasia of one or more digits or toes may be seen. There may be an associated camptodactyly (permanent flexion) or clinodactyly (abnormally positioning) of the fifth finger.
Neurological problems are less common and are said to occur in 30% of affected families.1 The clinical expression varies widely within and between affected families, as does the age of onset. Neurological involvement is usually evident by the second decade of life but may occur much later. Slowly progressive spastic paraparesis is the most common feature and is associated with characteristic brain MRI changes, described below. Other variable manifestations include neurogenic bladder and bowel disturbance, ataxia, dysarthria, seizures, and mild mental retardation. Neuro-ophthalmological findings include ptosis, nystagmus, gaze palsies, squint and visual impairment that is probably related to glaucoma or amblyopia.2 No neuropathological postmortem findings have been reported yet in these patients.2
Less common features include hypotrichosis (poor hair growth in 26%), brittle nails, microcephaly, and cleft palate. A few affected individuals have palmoplantar keratoderma (abnormal thickening of the palms and soles), dysplastic ears, conductive hearing loss, and cardiac anomalies, including arrhythmias or congenital malformations (ventricular septal defect).
The typical MRI changes are bilateral hyperintensity in the white matter in T2-weighted images involving the periventricular parieto-occipital region, and extending into the posterior limbs of the internal capsules and along the corticospinal tracts. It has been suggested that these changes are associated with the clinical neurological manifestations of ODDD, and their severity may be reflected in the phenotype.2 Other MRI findings include signal hypointensity of the globus pallidus, thalamus and cortex, which may be due to premature iron deposition.2 The Spinal cord images may be normal or show mild atrophy.
Although ODDD mutations have a high penetrance, they exhibit great intra- and inter- familial phenotypic variability that is not related to the mode of inheritance or the mutation type, but to variable expression of the GJA1 gene.1 GJA1 encodes connexin 43, which is one of 21 connexin proteins that participate in the formation and maintenance of intercellular channels.1 Each of these proteins affects different functional properties of the channel, including pore conductance, size selectivity, charge selectivity, voltage gating, and chemical gating, influencing the exchange of small ions and signaling molecules between cells.
Early recognition of the syndrome is important for the prevention and treatment of the clinical manifestations. Management is multidisciplinary. Clinicians should be alert to the possibility of ophthalmic, audiological, neurological, and dental complications in particular. Drugs that may precipitate glaucoma should be avoided. Plastic or orthopedic surgery is indicated for severe limb malformations. Genetic counseling should be offered and prenatal mutational analysis may be considered.
References
- Paznekas WA, Karczeski B, Vermeer S, et al. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat. 2009;30:724-33.
- Loddenkemper T, Grote K, Evers S, et al. Neurological manifestations of the oculodentodigital dysplasia syndrome. J Neurol. 2002;249:584-95.