
Summary
- Hearing impairment is very common and highly heterogeneous.
- Both environmental and genetic factors contribute to deafness.
- Progressive hearing loss is a major target for development of treatments.
- Likely vulnerable sites in progressive hearing loss include: the hair bundle of sensory hair cells; synapses below inner hair cells; and maintenance of cochlear homeostasis by the lateral wall of the cochlear duct.
- Mouse mutants can be used as tools to understand the biological mechanisms and molecular networks underlying human deafness.
Clues to the causes of deafness
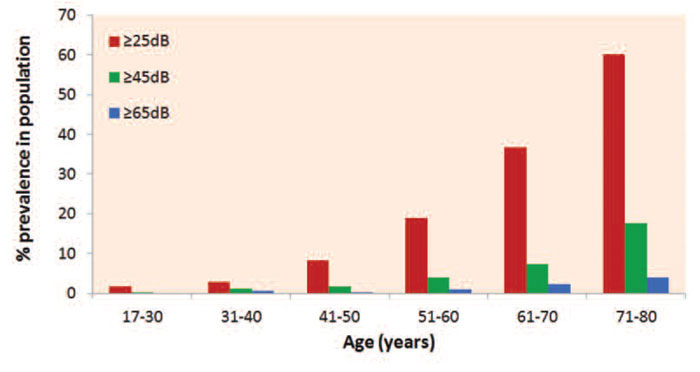
Hearing impairment is the most common sensory disorder in the human population and progressive hearing loss is particularly common. Around 1 in 850 children are born with a significant hearing impairment, and by the end of the first decade of life the number affected is double suggesting a considerable amount of early-onset progressive hearing loss.1 With each successive decade, more people are affected until over half of adults in the over 70 age group have a hearing loss of 25dB or greater2 (Figure 1). Despite the clear impact of early developmental defects of the ear on individuals and their families, the majority of people affected by deafness suffer progressive hearing loss, so this is the major problem to be solved. Hearing loss is profoundly isolating, both socially and economically, and has a major impact on the quality of life of those affected. The vast majority of affected individuals have no molecular diagnosis. This is especially true in cases of later-onset, age-related progressive hearing loss.
What do we know about the causes of hearing loss?
Hearing impairment is a heterogeneous disorder with a wide range of causes. We know that noise, drugs, infection and other external factors can trigger hearing loss, but there is a strong genetic component too. It is generally thought that half of all cases of permanent childhood deafness are due to single gene mutations, but this may well be an underestimate. Studies of childhood deafness show that GJB2 (encoding Connexin 26) is the most frequently associated gene. For example, in the UK, GJB2 mutations account for one third of cases where there are at least two affected sibs in a family, indicative of a recessive genetic cause.3 Another recent study of a US patient group (including apparently recessive, dominant and sporadic cases) using massively parallel sequencing of genes known to underlie deafness found 6% of cases were due to GJB2 mutations.4 Other genes are involved in much smaller proportions of cases, with the specific genes involved varying between different populations.3,4 However, cases of childhood deafness with known genetic aetiology represent the minority of cases of hearing impairment (see Figure 1), and there is very little information available on the genetic basis of deafness in the population as a whole. While many people have a single gene mutation causing deafness, others have multiple genes acting together. Other people carry variants of genes which result in an increased susceptibility to damaging agents. The best known example of this is the A1555G mutation of the mitochondrial genome, which confers extreme sensitivity to the ototoxic effects of aminoglycoside drugs to carriers,5 but there will be many further examples where single gene variants or collections of variants in multiple genes (genetic background) influence the susceptibility of a person to hearing loss following an environmental insult. It is difficult to identify these variants directly in humans because of the vast number of differences in the genome amongst individuals, but mice with a uniform genetic background and a mutation affecting a single gene can be used to dissect the underlying molecular basis of deafness. In this respect, genetics, and mouse genetics in particular, can be used as a tool to find genes and proteins associated with hearing ability, and discovery of these genes will be valuable in understanding the molecular processes underlying hearing loss whatever the trigger.
The research community has been remarkably successful in identifying genes underlying childhood deafness, including both syndromic forms and non-syndromic deafness. In the latter case, there are few clues to distinguish different types of hearing impairment. For this reason, gene identification has often relied upon tracking the inheritance of mutations in very large families or population isolates where it is likely that a single mutation is involved. Since the first gene (GJB2) involved in non-syndromic deafness was identified in 1997 by Kelsell and colleagues,6 65 genes have been discovered with mutations causing uncomplicated hearing impairment and a further 48 chromosomal loci have been reported to harbour mutations leading to non-syndromic deafness. Up-to-date details of these are listed on the Hereditary Hearing Loss website.7 There are many additional genes associated with syndromic deafness.8 Thus, there are likely to be several hundred genes implicated in human hearing impairment.
The types of genes involved in deafness are varied, covering a wide range of functions from transcription factors, extracellular matrix proteins, ion channels and transporters, junctional proteins, a range of unconventional myosins and associated scaffolding proteins, and molecules involved in synaptic function.9 Given the complex and highly specialised nature of the auditory system and its exquisite organisation, able to detect extremely small movements of less than a nanometer presented at rates of up to 100,000 times per second, it is not surprising that many different genes are involved.
Within the cochlea, the organ of Corti contains sensory hair cells that detect the mechanical stimulus of sound through the deflection of the array of stereocilia (modified microvilli) on their upper surface, which in turn puts tension on fine links between adjacent stereocilia directly opening the transduction channel (Figure 2). Cations flood through the open channel and depolarise the hair cell, initiating synaptic activity at the base of the cell. The rapid entry of cations is driven by a strong electrochemical gradient as the stereocilia are bathed by endolymph, an unusual extracellular fluid rich in potassium and maintained at a resting potential of around +100mV, while the internal voltage of the hair cell is around -60mV. Hair cells are surrounded by a set of highly differentiated supporting cells, some of which appear specialised to hold the hair cells rigidly in place while others express genes suggesting a role in homeostasis (Figure 2b). Inner hair cells are the primary receptor cells, receiving most of the afferent innervation. Outer hair cells respond to depolarisation by changing their length, boosting the motion of the organ of Corti and thus acting as amplifiers of the mechanical stimulus delivered to the inner hair cells.
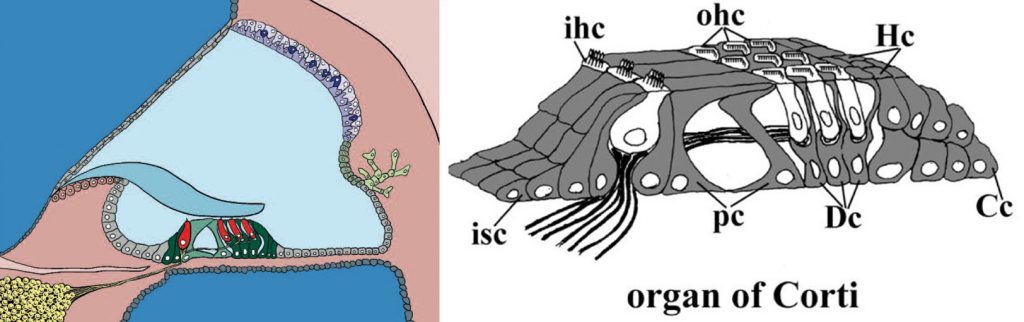
Figure 2B (right) The organ of Corti in detail, showing inner hair cells (ihc), outer hair cells (ohc) and several of the specialised types of supporting cells: pillar cells (pc), Deiters’ cells (Dc), Claudius cells (Cc), Hensen’s cells (Hc), and inner sulcus cells (isc).
Figure 2A was drawn by Morag Lewis, and 2B by Amy Kiern
Unlike mechanosensory epithelia in other vertebrates, the adult mammalian organ of Corti is unable to regenerate hair cells, so when a hair cell is damaged and dies, it is never replaced. The inability to regenerate hair cells may be a result of the extreme structural and functional specialisation of the mammalian cochlea. However, hair cell death is rarely if ever the initial cause of hearing impairment; hair cell death is preceded by dysfunction either of the hair cell itself or associated homeostatic processes.
The mechanisms underlying hearing impairment are as varied as the genes involved, and the mouse has played a key role in understanding these mechanisms. Any part of the auditory pathway can be involved from the external ear, through the middle ear and inner ear to the central auditory nuclei, although there are very few examples of deafness caused by primary central problems. One surprise has been the discovery of nearly 50 single genes in which mutations predispose to otitis media in the mouse.10 However, the majority of cases of hearing impairment in humans and the majority of single gene mutations causing deafness in the mouse are associated with primary dysfunction of some part of the cochlea. This dysfunction can result from early developmental defects of the inner ear giving congenital or early-onset deafness, or the dysfunction can have a later onset leading to progressive hearing loss. Judging from what we know about mechanisms involved in progressive hearing loss in animal models, there appear to be three main sites within the cochlea that are particularly vulnerable to pathological processes.
Firstly, the precise arrays of stereocilia (hair bundles) at the top of hair cells are often damaged following excessive noise exposure,11 and many different genes are required for their normal development and function (e.g. Myo7a, ref 12; Cdh23, ref 13). Stereocilia can be too long or too short, thin, disorganised, misoriented, collapsed, splayed, or fused together in different mouse mutants. Given the role of the hair bundles in auditory transduction and the nature of the fine links between them that are essential for function, it would not be surprising if they were readily damaged in progressive hearing loss.
Secondly, synapses below inner hair cells appear to be particularly vulnerable to damage. Levels of noise exposure that were thought to produce only reversible, temporary shifts in auditory thresholds actually lead to permanent damage.14-16 Afferent fibres with high thresholds and low spontaneous rates are selectively lost, and these are thought to have a role in detecting sounds in noisy backgrounds, a feature that is often an early sign of hearing loss in humans. Furthermore, a common consequence of exposure to damage is swelling of the afferent nerve endings below inner hair cells, a classic indicator of glutamate excitotoxicity, again indicating this is an area of vulnerability.17-18
Thirdly, the lateral wall of the cochlear duct (Figure 2a) has a key role in controlling homeostasis of the local environment of the organ of Corti, including pumping potassium into the endolymph at such a rate that its resting potential is maintained at its normal high level of +100mV. This is a highly energy-dependent process, requiring an ample blood supply as found in the stria vascularis on the lateral wall opposite the organ of Corti. Reduced potentials have been reported in some models of progressive hearing loss.19 Signs of deficient fluid homeostasis have been found in human temporal bone specimens, leading to the proposal of a separate pathological process called strial or metabolic hearing loss in progressive or age-related deafness.20
Mouse mutants with each of these types of pathology are available to provide clues to the biological mechanisms and molecular networks involved. Using mouse models allows us to develop a greater understanding of auditory function and pathology which is transferable to humans. Until we have a molecular understanding of the processes underlying progressive hearing loss, it will be difficult to develop treatments. The only remedial options commonly available are hearing aids and cochlear implants, prosthetic devices that provide benefits but do not restore normal function. There is an unmet need for medical approaches to slow down or reverse progressive hearing loss, and the challenge now is to use the new genetic resources available to build that understanding and develop new treatments.
References
- Fortnum HM, Summerfield AQ, Marshall DH, Davis AC, Bamford JM. Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. British Medical Journal 2001;323:536-40.
- Davis AC (1995) Hearing in Adults. London: Whurr.
- Hutchin T, Coy NN, Conlon H, Telford E, Bromelow K, Blaydon D, Taylor G, Coghill E, Brown S, Trembath R, Liu XZ, Bitner-Glindzicz M, Mueller R. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK – implications for genetic testing. Clin Genet 2005;68:506-12.
- Shearer AE, Black-Ziegelbein EA, Hildebrand MS, Eppsteiner RW, Ravi H, Joshi S, Guiffre AC, Sloan CM, Happe S, Howard SD, Novak B, Deluca AP, Taylor KR, Scheetz TE, Braun TA, Casavant TL, Kimberling WJ, Leproust EM, Smith RJ. Advancing genetic testing for deafness with genomic technology. J Med Genet. 2013;50:627-34.
- Prezant TR, Agapian JV, Bohlman MC, Bu X, Öztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, Shohat M, Fischel-Ghodsian N. Mitochondrial ribosomal RNA mutation associated with both antibiotic−induced and non−syndromic deafness. Nature Genetics 1993;4:289-94.
- Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997;387:80-3.
- Van Camp G, Smith RJH. Hereditary Hearing Loss Homepage. 2013; http://hereditaryhearingloss.org
- Online Mendelian Inheritance in Man, OMIM®. 2013; http://omim.org/
- Dror AA, Avraham KB. Hearing impairment: a panoply of genes and functions. Neuron 2010;68:293-308.
- Tyrer HE, Crompton M, Bhutta MF. What Have We Learned from Murine Models of Otitis Media? Curr Allergy Asthma Rep. 2013;PMID:23775349 (in press).
- Wang Y, Hirose K, Liberman MC. Dynamics of noise-induced cellular injury and repair in the mouse cochlea. J Assoc Res Otolaryngol. 2002;3:248-68.
- Gibson F, Walsh J, Mburu P, Varela A, Brown KA, Antonio M, Beisel KW, Steel KP, Brown SD. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature 1995;374:62-4.
- Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27:103-7.
- Kujawa SG, Liberman MC. Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. J Neurosci. 2006;26:2115-23.
- Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci. 2009;29:14077-85.
- Furman AC, Kujawa SG, Liberman MC. Noise-induced cochlear neuropathy is selective for fibers with low spontaneous rates. J Neurophysiol. 2013;110:577-86.
- Pujol R, Puel JL, Gervais d’Aldin C, Eybalin M. Pathophysiology of the glutamatergic synapses in the cochlea. Acta Otolaryngol. 1993;113:330-4.
- Robertson D. Functional significance of dendritic swelling after loud sounds in the guinea pig cochlea. Hear Res. 1983;9:263-78.
- Schulte BA, Schmiedt RA. Lateral wall Na,K-ATPase and endocochlear potentials decline with age in quiet-reared gerbils. Hear Res 1992;61:35–46.
- Schuknecht HF, Gacek MR. Cochlear pathology in presbycusis. Ann Otol Rhinol Laryngol. 1993;102:1-16.