

Abstract
Huntington disease (HD) has a prolonged premanifest phase. Detailed premanifest HD studies followed identification of the causal CAG repeat expansion in the Huntingtin gene in 1993 that allowed genetic testing. A better understanding of the years before clinical diagnosis and variation in disease presentations resulted. Information from these premanifest studies and new biomarkers may enable a wider definition of HD, earlier diagnosis and care, as well as better measures of progression in clinical trials.
Assessments
A detailed history, from the person and a companion is important to determine the full extent of the disease features, family history, social support network, level of function and impact on the person and those around them. The Unified Huntington Disease Rating Scale (UHDRS) documents cognitive, behavioural, motor and functional manifestations. This validated scale is used in most HD observational and drug trials. A diagnosis is made based on the DCL (diagnostic confidence level) rated 0-4, with a score of 4 equating to a > 99% certainty that the motor findings are due to HD. There is some subjectivity to this decision made after the motor examination, documented as the UHDRS total motor score (TMS). This scale scores the eye movement disorder, speech, finger taps, tandem gait, chorea and other motor features.4 Cognitive testing is essential at presentation. The Montreal Cognitive Assessment is preferred over the MMSE. Dysphagia, weight loss, impulsiveness and unawareness, although prominent, are not directly included in the UHDRS.1
Presentations of manifest HD and the variable phenotype
Patients who present to the clinic, especially with an unknown family history or a phenotypic variation, may have well-established disease. Disease features of cognitive impairment and unawareness contribute to presentations later in the disease course.5 Younger onset (Juvenile) with a longer repeat expansion,6 late onset,7 and those with predominantly cognitive and behavioural manifestations, occur.8 The main reason for the different ages of onset is variation in the length of the CAG repeat expansion within the HTT gene.1 The longer the repeat, the earlier the onset. Variations in onset age and features, even with the same repeat length, are postulated to be due to environmental and genetic modifiers.9
The contribution of premanifest studies
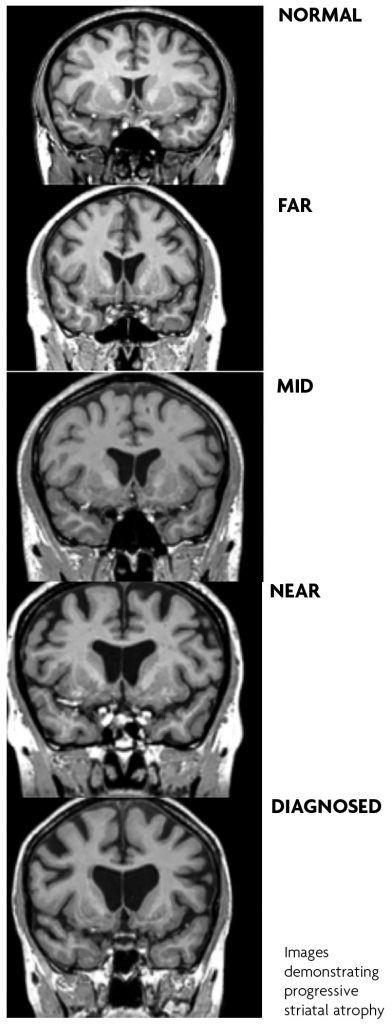
HD is a disease with a single genetic cause with established genetic testing programmes for 25 years. A number of premanifest studies of mutation carriers have contributed to our knowledge of this phase of the condition. Amongst the most detailed are the PREDICT-HD study of over 1000 individuals with the repeat expansion that ran over 10 years,10 the PHAROS study11 of 983 untested individuals ‘at risk’ because of family history and the TRACK HD study.12 In addition, large longitudinal observational studies including COHORT, Registry and the current ENROLL study, added even more details about the premanifest path to clinical diagnosis and progression.1,9 The major finding from these studies is that subtle but progressive changes in neuroimaging findings, particularly striatal volumes, cognition, behaviour and motor examination occur during the premanifest phase and progress over the years before a definite clinical diagnosis. These findings are sufficient to define a prodromal phase closer i.e. within five years, of clinical onset.
With more experience of this crossover from premanifest to definite disease, the diagnosis can be made earlier than previously. Although changes occur in domains other than motor, most would not be confident to make a diagnosis of HD based on behavioural and cognitive features alone.
Research diagnosis
As in other dementias, most notably Alzheimer’s disease, a clinical diagnosis is separated from the research diagnosis.
Genetic testing for the CAG expansion is available to those aged 18 years and over. Many know of their carrier status for a long period before their eventual disease onset and perhaps witnessed it in their affected relatives.
These premanifest individuals usually present earlier for clinical diagnosis. Some seek reassurance that overt disease is not yet evident. Some decide on testing and then prefer no service contact and to live life as normal until affected, often significantly. A greater proportion of unaffected people with a family history, decide not to have a premanifest genetic test.
Predictions of onset have been extrapolated from the CAG repeat lengths. It is not possible to determine age of onset accurately for an individual based on the CAG repeat length. It is however possible to tell an 18 year old with a repeat length of 40 that they will not be affected for some years i.e. 10 or more likely 20 years.
Progressive atrophy notably in the striatum, corpus callosum, insular in particular are documented on a range of imaging modalities in the premanifest phase before overt signs of disease.10,12
Measurement of Huntingtin protein in the CSF and blood and more recently in saliva is possible.
Manifest disease would not be diagnosed in the clinic based on these earliest changes, although these components are valuable in research studies, if standardised and reproducible as possible biomarkers of progression.
What are the premanifest and clinical findings that assist in diagnosis?
On a recent review of data from HD studies, TMS, Total Functional Capacity (TFC), Symbol Digit Modality and Stroop Word Reading are considered the most reliable items to support the clinical diagnosis in this earliest stage of premanifest to manifest diagnosis. As a result, Schobel et al propose a composite UHDRS.13
Why a diagnosis?
With genetic testing, protocols became available to assist in ensuring fully informed consent before testing premanifest, unaffected people. The process of testing, the reaction to results and confidentiality and discrimination concerns are emphasised. Diagnostic testing for people with a suspected clinical diagnosis of HD is a confirmatory test. An explanation of the implications of a positive test in this situation should be undertaken and where possible consent of the person or a person responsible is required.
There is no cure. Although there are recent promising advances in genetic therapies14 that target the expanded repeat, a cure is some years ahead.
The premanifest group, often years away from clinical onset, face uncertainty about the time of definite diagnosis. The task of deciding whether symptoms and signs in these people constitute a clinical diagnosis and when to disclose a definite diagnosis can be difficult.15 Many manifestations found in the premanifest study participants, occur in the unaffected population e.g. depression, apathy and irritability. In addition unawareness of disease onset is prominent in HD including at an early stage. Sometimes this is because the signs visible on examination do not produce any functional impact i.e. the eye movement disorder of slow pursuit and saccades. Very obvious chorea may not be noticed and along with the behavioural changes are often more apparent to a companion. Studies show that the divergent reports of symptoms and signs between the person, their companion and the examiner, are consistent with unawareness.5
It could be argued that if the person is unaware of manifestations and there is no functional impact, then why make an earlier diagnosis? This raises many valid ethical considerations, not the least being the person’s right to know.
It is important that disclosure of a changed status from premanifest to manifest is undertaken with care and based on a reliable, accurate history and reproducible signs and after assessing available support systems.
Beyond diagnosis
At any time, but including when premanifest “conversion” to manifest HD is disclosed, more is required than ‘just a diagnosis’. An early follow up, ideally to an easily accessible, knowledgeable service with multidisciplinary care, should be offered. Ongoing contact with a general practitioner for extra support and to maintain good general health is advised and hopeful but realistic discussion of research advances.
As with other diseases of the nervous system, a healthy brain/life style intervention is recommended and cognitive and physical activity. Limiting other factors that affect the brain, i.e. alcohol and drug abuse and smoking is important as well as managing co-morbidities, including hypertension, diabetes and hypercholesterolaemia. The cognitive/behavioural and psychiatric manifestations may be prominent.16 Many are treatable. Emphasis on review for these features is recommended and early intervention. Preparation for the future should be advised but the slow course emphasised. HD runs a course of up to 20 years with considerable variation. Patient and carer education about manifestations, including unawareness and the other non-motor manifestations and support from disease societies and initiatives helps. Today’s premanifest generations have a reasonable expectation that their outcome and course will differ significantly from that of their affected family members.
References
- Ghosh R, Tabrizi SJ. Huntington disease. Handb Clin Neurol. 2018;147:255-278.
- Sachdev PS, Blacker D, Blazer DG et al. Classifying neurocognitive disorders: the DSM-5 approach. Nat Rev Neurol. 2014 Nov;10(11):634-42.
- Rawlins MD, Wexler NS, Wexler AR, Tabrizi SJ et al.The Prevalence of Huntington’s Disease. Neuroepidemiology. 2016;46(2):144-53.
- Unified Huntington’s Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Disord. 1996 Mar;11(2):136-42.
- Sitek EJ, Thompson JC, Craufurd D, Snowden JS. Unawareness of deficits in Huntington’s disease. J Huntingtons Dis. 2014;3(2):125-35.
- Moser AD, Epping E, Espe-Pfeifer P, Martin E et al. A survey-based study identifies common but unrecognized symptoms in a large series of juvenile Huntington’s disease. Neurodegener Dis Manag. 2017 Oct;7(5):307-315.
- Chaganti SS, McCusker EA, Loy CT. What do we know about Late Onset Huntington’s Disease? J Huntingtons Dis. 2017;6(2):95-103.
- Paulsen JS, Long JD. Onset of Huntington’s disease: can it be purely cognitive? Mov Disord. 2014 Sep 15;29(11):1342- 50.
- Lee JM, Ramos EM, Lee JH, Gillis T et al and Paulsen JS; PREDICT-HD study of the Huntington Study Group (HSG), Landwehrmeyer GB; REGISTRY study of the European Huntington’s Disease Network, Myers RH; HD-MAPS Study Group, MacDonald ME, Gusella JF; COHORT study of the HSG. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012 Mar 6;78(10):690-5.
- Paulsen JS, Long JD, Ross CA, Harrington DL et al and PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Prediction of manifest Huntington’s disease with clinical and imaging measures: a prospective observational study. Lancet Neurol. 2014 Dec;13(12):1193-201.
- Huntington Study Group PHAROS Investigators, Biglan KM, Shoulson I, Kieburtz K, Oakes D et al Clinical- Genetic Associations in the Prospective Huntington at Risk Observational Study (PHAROS): Implications for Clinical Trials. JAMA Neurol. 2016 Jan;73(1):102-10.
- Tabrizi SJ, Scahill RI, Owen G, Durr A et al and TRACK-HD Investigators. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013 Jul;12(7):637-49.
- Schobel SA, Palermo G, Auinger P, Long JD et al and TRACK-HD, COHORT, CARE-HD, and 2CARE Huntington Study Group Investigators. Motor, cognitive, and functional declines contribute to a single progressive factor in early HD. Neurology. 2017 Dec 12;89(24):2495-2502.
- Wild EJ, Tabrizi SJ. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017 Oct;16(10):837- 847.
- McCusker EA, Loy CT. Huntington Disease: The Complexities of Making and Disclosing a Clinical Diagnosis After Premanifest Genetic Testing. Tremor Other Hyperkinet Mov (N Y). 2017 Sep 6;7:467.
- Epping EA, Kim JI, Craufurd D, Brashers-Krug TM et al and PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Longitudinal Psychiatric Symptoms in Prodromal Huntington’s Disease: A Decade of Data. Am J Psychiatry. 2016 Feb 1;173(2):184-92. Epub 2015 Oct 16.