


Summary
- Mitochondrial dysfunction is a pathogenic mechanism in Parkinson’s disease.
- Mitochondrial dynamics (refers to fission, fusion and movement of mitochondria) can highly affect mitochondrial function and hence, neuronal activity and viability.
- In recent years imbalances in mitochondrial dynamics have been reported in a wide range of experimental models of Parkinson’s disease. Targeting this pathway has emerged as a potential novel therapeutic avenue for Parkinson’s disease.
Parkinson’s disease
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder. Motor features such as resting tremor, bradykinesia, rigidity and postural instability, can be attributed to the degeneration of dopaminergic neurons in the substantia nigra pars compacta. Because extra-nigrostriatal regions are also affected in PD, non-motor symptoms, including but not limited to depression, cognitive deficits and dementia, and some autonomic impairments are also commonly observed in these patients. Together, these motor and non-motor features cause significant disability and drastically reduce quality of life in the afflicted patients.
It has been estimated that up to 10 million people worldwide are affected by PD and approximately 100,000 people in the UK are living with this disease.1 This prevalence will increase dramatically with the ageing population. PD also causes an enormous economic burden. Combining both the direct costs (medication and healthcare resources use) and indirect costs (loss of productivity, unemployment, and mortality costs), PD is estimated to have a staggering economic burden of £3.3 billion annually in the UK.2 Additional effective treatments for this devastating disease are urgently needed. To achieve this goal, it is critical to understand the aetiology and underlying mechanisms of neuronal dysfunction and degeneration in PD.
The cause(s) of the majority of PD cases remains unknown. Epidemiological studies indicate that both the environment and genetic susceptibility most likely play a role in sporadic PD. Significant strides, however, have been made in familial PD studies in the past decade. Although currently less than 10% of PD cases can be directly linked to monogenic mutations, studies from these autosomal dominant (SNCA, LRRK2) and recessive (Parkin, DJ1, PINK1 and ATP13A2) genes have provided significant insights into potential mechanisms of neuronal dysfunction and degeneration in PD. These non-mutually exclusive mechanisms include mitochondrial dysfunction, oxidative stress, neuroinflammation, protein misfolding, and insufficient autophagic or proteasomal protein degradation.
Mitochondrial dysfunction in PD
The initial proposal of mitochondrial dysfunction as a pathogenic mechanism in PD originated from the discoveries that 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced parkinsonism in humans and subsequently in various laboratory animal species. Because the active metabolite of MPTP blocks the mitochondrial complex I function, many investigators have actively searched for mitochondrial defects in PD patients and developed therapeutic strategies targeting the electron transport chain. However, accumulating studies from recent years show that other innovative strategies aimed at restoring mitochondrial function have considerable beneficial potential with wide applicability to other diseases. These studies, led initially by the observations of mutations in PTEN-induced putative kinase-1 (PINK1) and parkin, not only have affirmed the critical role of mitochondria in PD but also have uncovered mitochondrial dynamics (fission / fusion / movement) as a potential therapeutic target for PD.3,4
Impact of mitochondrial dynamics on neuronal function and survival
Mitochondria are “dynamic” organelles. They constantly undergo changes in shape, size, number and location. These alterations can be affected by mitochondrial morphology, which in turn, is controlled mainly by the processes of fission and fusion. As illustrated in Figure 1, in mammals, the outer mitochondrial proteins mitofusins (Mfn1 and Mfn2) and the inner mitochondrial protein optic atrophy 1 (OPA1) are responsible for mitochondrial fusion. Fission on the other hand requires the recruitment of dynamin-related protein 1 (Drp1) from the cytosol to the outer mitochondrial membrane by Fission-1 (Fis1) and mitochondrial fission factor (Mff).
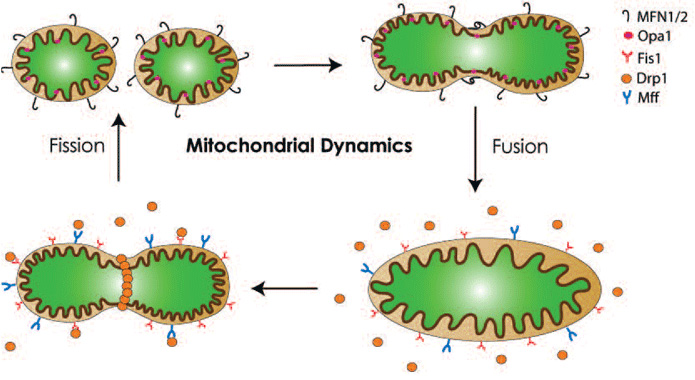
Fission produces multiple smaller mitochondria which are more motile within the cell and therefore facilitating mitochondrial trafficking to neuronal dendrites and axons.5 Fission is also important for segregating dysfunctional mitochondria from healthy counterparts.6 Through this segregation, damaged mitochondria are subsequently removed by autophagy (known as mitophagy). Fission, therefore, also plays a role in quality control for the maintenance of a healthy pool of mitochondria. However, as discussed later, the in vivo significance of this mechanism remains to be established. In contrast to fission, fusion results in larger and highly interconnected networks of mitochondria, which could offer a larger ATP supply. Furthermore, it has been established that within a single cell, wild type mitochondrial DNA (mtDNA) and mutant variants can co-exist (a scenario known as heteroplasmy) and when the damaged load exceeds a threshold of >60%, mitochondrial dysfunction occurs.7 By promoting exchanges of mtDNA between functional and defective mitochondria in heteroplasmic cells, mitochondrial fusion can dilute defective mitochondria and attenuate the negative impact of damaged mtDNA through functional complementation.3,8 Damages or mutations in mtDNA of nigral dopaminergic neurons have been detected in experimental models of PD as well as in patients with sporadic PD.7,9 Lastly, mitochondrial fusion prevents cell death by blocking the release of the pro-apoptotic protein cytochrome c.3 In short, a balance of fusion and fission is crucial not only to mitochondrial morphology, but also to function and survival of cells. Thus, it does not come as a surprise that imbalances in this pathway have been linked to various human diseases,10 including PD.
Potential clinical relevance of impaired mitochondrial dynamics in PD
Imbalances in mitochondrial fission/fusion have been linked to several prominent genetic mutations in PD. Mutations in LRRK2 are the most common cause of familial PD. In mammalian cell culture models, pathogenic LRRK2 mutations induce excessive mitochondrial fragmentation in a Drp1-dependent mechanism. Blocking mitochondrial fission or promoting mitochondrial fusion improves mitochondrial morphology, function and cell viability.11 SNCA, which encodes α-synuclein, is another prominent autosomal dominant PD gene. The discoveries of mutations in this gene have revolutionised PD research in the past two decades. Recent genome-wide association studies have identified both SNCA and LRRK2 as major genes linked to sporadic PD. Like LRRK2, α-synuclein has been reported in cultured cells to induce severe mitochondrial fragmentation.12-15 Although it is still a topic of debate regarding the mechanisms by which this excessive mitochondrial fission occurs, α-synuclein has been reported to increase Drp1 function13 and to reduce OPA1 expression,15 leading to an overall enhanced fission effect. Imbalanced mitochondrial fission/fusion has also been widely observed in recessive genes (PINK1, parkin and DJ1) causing PD. PINK1 and parkin are most extensively studied (see review16 for a more detailed discussion of various PINK1/parkin studies). Overall, based on most mammalian cell culture models, loss of PINK1 and parkin function has been demonstrated to tip the balance of mitochondrial fission/fusion to an overall enhanced pathogenic fission. Either blocking fission or promoting fusion confers protection in these studies. However, these results are not consistent primarily with those obtained from the drosophila models.16 In addition to PD genes, neurotoxic molecules capable of damaging the nigrostriatal pathway such as rotenone, MPTP, methamphetamine and 6-hydroxydopamine, have been reported to cause mitochondrial fission and neurotoxicity that can be attenuated by promoting fusion or blocking fission. Perturbed mitochondrial dynamics may, therefore, represent a shared mechanism that underlies both genetic and toxin-induced related PD.
Conclusion
The link between mitochondrial dysfunction and PD has been proposed since the discovery of parkinsonism in humans caused by MPTP-contaminated synthetic meperidine in the early 1980s. Advances in the genetics of PD have now established a strong genetic link between this neurological disorder and mitochondria. Furthermore, these genetic studies have highlighted mitochondrial dynamics with a tremendous potential to restore mitochondrial function. Moving forward, to make this target one step closer to “from bench to bedside”, first, the in vivo significance of manipulating mitochondrial dynamics in animal models of PD must be vigorously evaluated. Second, given that both fission and fusion are important physiologically, it is critical to determine whether and to what extent mitochondrial fission or fusion should be promoted in PD. This answer should become clearer based on future studies of animal models of PD and brain samples of PD patients. Based on an animal study using a mouse model with a conditional deletion of Mfn2 in dopaminergic neurons,17 the loss of this mitochondrial fusion gene results in progressive retrograde neurodegeneration and L-DOPA responsive locomotor deficits. Simultaneous deletion of Mfn2 in dopaminergic neurons in the ventral tegmental area further reveals that nigral dopaminergic neurons are most vulnerable to enhanced mitochondrial fission. Combined with other in vitro studies, this animal study supports promoting mitochondrial fusion as a strategy for PD. One potential concern with this approach is that mitochondrial fission is important for mitophagy. However, a recent study points out that mitophagy might not occur in the mammalian brain.18 In short, manipulating mitochondrial dynamics pathway, perhaps by blocking mitochondrial fission or promoting fusion, represents a high potential for novel therapeutic strategy for PD.
References
- Dorsey, ER, Constantinescu, R, Thompson, JP, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68:384-6.
- Findley, LJ. The economic impact of Parkinson’s disease. Parkinsonism Relat Disord. 2007;13 Suppl:S8-S12.
- Youle, RJ and van der Bliek, AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062-5.
- Andreux, PA, Houtkooper, RH, and Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov. 2013;12:465-83.
- Detmer, SA and Chan, DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870-9.
- Twig, G, Elorza, A, Molina, AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433-46.
- Bender, A, Krishnan, KJ, Morris, CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515-17.
- Chen, H and Chan, DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci. 2010;1201:21-5.
- Kraytsberg, Y, Kudryavtseva, E, McKee, AC, et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518-20.
- Archer, SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236-51.
- Wang, X, Yan, MH, Fujioka, H, et al. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. 2012;21:1931-44.
- Kamp, F, Exner, N, Lutz, AK, et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29:3571-89.
- Gui, YX, Wang, XY, Kang, WY, et al. Extracellular signal-regulated kinase is involved in alpha-synuclein-induced mitochondrial dynamic disorders by regulating dynamin-like protein 1. Neurobiol Aging. 2012;33:2841-54.
- Nakamura, K, Nemani, VM, Azarbal, F, et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem.2011;286:20710-26.
- Guardia-Laguarta, C, Area-Gomez, E, Rub, C, et al. alpha-Synuclein Is Localized to Mitochondria-Associated ER Membranes. J Neurosci. 2014;34:249-59.
- Exner, N, Lutz, AK, Haass, C, et al. Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012;31:3038-62.
- Pham, AH, Meng, S, Chu, QN, et al. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum Mol Genet. 2012;21:4817-26.
- Sterky, FH, Lee, S, Wibom, R, et al. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A. 2011;108:12937-42.