

For some years disease modifying treatments (DMTs) for relapsing remitting multiple sclerosis (RRMS) have been limited to ‘injectable’ therapies consisting of just two classes of drugs, beta interferon (IFN-b) and glatiramer acetate (GA). It is recognised that IFN-b and GA have limited efficacy and tolerability.1-2 The development of natalizumab has provided a treatment of greater efficacy but by NICE criteria is limited to those with rapidly evolving severe relapsing remitting multiple sclerosis (RES RRMS).3 There are now three oral DMTs for RRMS licenced by the European Medical Agency (EMA), two with full published NICE guidance (fingolimod and teriflunomide), and one (dimethyl fumarate [DMF]) anticipated in summer of 2014. In addition, in the new NHS structure, specialist prescribing for MS is currently the remit of NHS England and therefore more specific guidance has been seen and is likely to come from this body.
Aside from the obvious attraction of a ‘pill’ for MS therapy, the increase in the options of DMTs, with different mechanisms of action and ranges of efficacy and side effects, all help in the goal of successful MS disease suppression. Clinicians now need to evaluate licencing, regulatory guidance, safety, side effects, and efficacy of the various DMTs available. This is the first of three articles which review the oral therapies, fingolimod, teriflunomide and DMF, to help the Neurologist guide patients through the ‘tyranny of choice’. (See article 2 on Teriflunomide here, and article 3 on Tecfidera here).
Fingolimod
Current Approval
Fingolimod (GilenyaTM) became the first oral therapy to be approved for the treatment of RRMS in 2010 by the USA. The European community following suit in 2011 when the EMA recommended fingolimod in patients with high disease activity despite IFN-b therapy, or rapidly evolving severe RRMS (RES RRMS). As of April 2012, NICE have recommended fingolimod for adults with RRMS who have been exposed to IFN-b with unchanged or ongoing relapse activity.4 NHS England has now clarified that GA (CopaxoneTM) is considered an equivalent of IFN-b and therefore patients who have had an adequate course of GA, but on-going disease activity are eligible for fingolimod, thus resolving an outstanding discrepancy. Our understanding is the EMA licence for fingolimod will be amended to be patients with high disease activity despite full and adequate treatment (considered 12 months of treatment) with at least one disease modifying therapy, ie those who have had at least a single relapse in 12 months whilst on any DMT and with 9 T2 lesions (in total) or 1 or more new gadolinium enhancing lesions.
More recently NHS England has provided criteria of positive JC virus serology, previous immunosuppressant therapy or two years of natalizumab therapy as justification for switching from natalizumab to fingolimod.
Fingolimod is taken as single tablet once daily at a dose of 0.5mg.
Mechanism of Action
Fingolimod is a pro-drug that is rapidly phosphorylated by sphingosine kinase 2 to the active metabolite sphingosine phosphate. Sphingosine phosphate is a functional antagonist of the sphingosine 1–phosphate (S1P) receptor subtypes 1, 3, 4 and 5 that are present on lymphocytes and various cells types of the CNS and other tissues.5 The mechanism of action of fingolimod is believed to be two-fold. Firstly it inhibits the ability of specific lymphocytes to leave lymph nodes causing a reduction in peripheral blood lymphocytes and their reduced migration into the CNS.6 Secondly, fingolimod is able to cross the BBB and may have direct effects on the CNS with evidence of reduced axonal damage, demyelination and astrogliosis.7
Fingolimod is absorbed slowly, taking 12-16 hours to reach a maximum concentration and the half-life is 6 to 9 days.8 The bioavailability is unaffected by ingestion of food.9 Fingolimod is primarily metabolised by cytochrome (CYP)4F2 but also via reversible phosphorylation and transformation by dihydroceramide synthase.10 Elimination is predominately via excretion of inactive metabolites in the urine (81%) with 2.5% of fingolimod and fingolimod phosphate excreted in the faeces.8
Clinical Efficacy
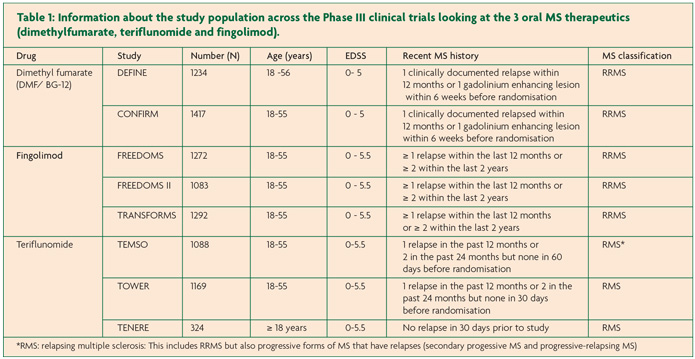
FREEDOMS, FREEDOMS II and TRANSFORMS are the pivotal Phase III trials that investigated the clinical efficacy of fingolimod.11-13 All three trials included over 1000 patients and were multi-centre, double-blind and randomised. FREEDOMS and FREEDOMS II had placebo arms and a study duration of 24 months whilst TRANSFORMS had IFN-b once weekly 30µg intramuscular (IM) injections (AVONEX™) as an active comparator and was shorter at 12 months. In all three trials fingolimod was given as a once daily dose of 0.5mg or 1.25mg however during the FREEDOMS II study, the participants in the 1.25mg arm were incorporated into the 0.5mg arm following a risk-benefit analysis. Only data for the licensed 0.5mg dose will be reviewed here (Table 2).
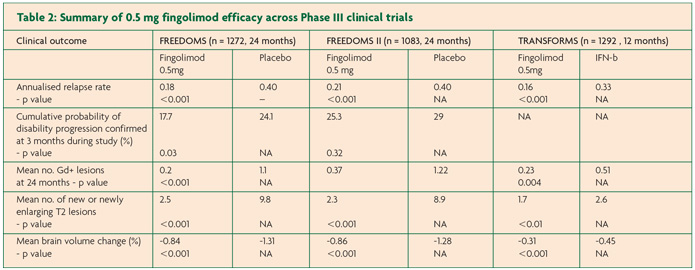
The primary end point of FREEDOMS, ARR, was significantly reduced by 54% for the fingolimod group compared to placebo (0.18 versus 0.40, p < 0.001).11 Fingolimod also reduced the risk of disability progression confirmed at three months by 30% and at six months by 37%. MRI investigations performed at 24 months showed 82% fewer gadolinium-enhancing lesions compared to the placebo arm (0.2 versus 1.1, p < 0.001) with a greater proportion of patients free from gadolinium-enhancing lesions in the fingolimod arm (89.7% versus 65.1%). There were also fewer new or enlarging T2 lesions (2.5 versus 9.8, p <0.001) and again a greater proportion taking fingolimod showed no new or enlarging T2-weighted lesions at 24 months compared to those on placebo (50.5% versus 21.2%, p <0.001). The atrophy data for fingolimod was also very promising with 35% less brain atrophy compared to placebo over the course of the trial (-0.84% versus -1.31%, p <0.001).
FREEDOMS II incorporated safety recommendations made by the FDA following FREEDOMS.12 Again, fingolimod significantly reduced ARR compared to placebo, this time by 48% (0.21 versus 0.40, p <0.0001). Disappointingly there was no difference in the second primary endpoint of disability progression. Data from imaging showed that those in the fingolimod group had significantly fewer gadolinium-enhancing lesions compared to placebo (0.37 versus 1.22, p <0.0001) and this finding was mirrored in the number of new/ enlarging T2 lesions (2.3 versus 8.9, p <0.0001). A similar result as seen in FREEDOMS was found for brain volume loss, with 33% less brain atrophy in fingolimod arm compared to placebo (-0.858% versus -1.279%, p <0.0002).
In TRANSFORMS, ARR was reduced by 52% in those taking fingolimod compared to IFN-b (0.16 versus 0.33, p <0.001).13 As may be expected in such a short trial there was no statistically significant difference in disability progression over the course of the study between fingolimod and IFN-b. There was however a 35% reduction in new or enlarging T2 lesions on MRI compared to IFN-b (p=0.004) and fewer mean gadolinium enhancing lesions (0.23 versus 0.51, p < 0.01). Brain atrophy data was also favourable with 33% less brain volume loss compared to the IFN-b (-0.31 versus-0.45 respectively p < 0.001).
Safety
Fingolimod has been studied in four major clinical trials and combined with postmarketing surveillance this amounts to more than 135,800 patient years.14 There were three deaths in FREEDOMS, only one of which was in the fingolimod arm (unlicensed 1.25mg dose) due to suicide.11 Two deaths occurred in TRANSFORMS, again both in the 1.25mg group; the first from disseminated primary varicella zoster who was taking concommitant steroids and the second from herpes simplex encephalitis.13
Adverse events occurred at a similar frequency between the 0.5mg and placebo arms in FREEDOMS, whereas they were more commonly seen in those taking IFN-b in TRANSFORMS (91.6% versus 86%, IFN-b and fingolimod 0.5mg arms respectively). Serious adverse events interestingly were higher in those receiving placebo (13.4%), than fingolimod 0.5mg (10.1%) in FREEDOMS.11
Pooled analysis across all three studies showed a higher incidence of bradycardia in the fingolimod arm compared to placebo (1% versus 0.6%).15 Bradycardia generally began after the first hour of taking the first dose, reached a peak four to five hours later and was asymptomatic. It is known that sphingosine-1 phosphate has direct effects on the pacemaker activity of the sino-atrial node.16 Systolic blood pressure across FREEDOMS rose modestly by 1.9mmHg and diastolic by 0.7mmHg in those treated with 0.5mg fingolimod.11

Unsurprisingly peripheral blood tests showed leucopenia (primarily lymphopenia). This is reversible with return to pre-medication levels some 8 to 16 weeks after cessation of fingolimod. Herpes infections, bronchitis and pneumonia occured more frequently in those taking fingolimod across all major studies.15 However overall infection rates were similar, with infection occurring in 67.9% of those on placebo versus 65.1% of those taking fingolimod 0.5mg, with no single infection correlating with lymphocyte count.15,17
Macular oedema occurred in 0.4% on fingolimod 0.5mg compared to 0.1% for placebo, generally three to four months after starting treatment.15 This is likely to be due to endothelial effects compromising the blood-retina barrier.18 Those with a history of diabetes and or uveitis appear to be particularly at risk.19
Reduced pulmonary function has also been observed in those taking fingolimod, with an average reduction in FEV1 of 3.1% and DLCO of 3.8% after 24 months (compared to 2% and 2.7% respectively in placebo).19
As seen in the other treatments discussed here, fingolimod is also associated with dysfunctional liver enzymes with more frequent cases of three-fold increases in liver enzymes in those taking 0.5mg compared to placebo and IFN-b (8.3% versus 1.8% versus 2.9% respectively).19 Liver function tests appear to return to baseline following treatment discontinuation.
Animal data suggests that fingolimod is teratogenic. 219 pregnancies have been reported in participants of the trials with 8/67 live births born with congenital abnormalities.20 Fertile female patients should be advised to take effective contraception until two months after treatment termination due to the time taken to eliminate fingolimod from the body.
Adverse events leading to discontinuation of study drug in FREEDOMS were equal at 8% in both the 0.5mg and placebo arms.11 Data from TRANSFORMS also had similar results with only 5.6% of those taking 0.5mg fingolimod and 3.7% of those on IFN-b discontinuing due to adverse effects.13
References
- Shirani A, Zhao Y, Karim ME, et al. Association Between Use of Interferon Beta and Progression of Disability in Patients With Relapsing-Remitting Multiple Sclerosis. JAMA 2012;308(3):247-256
- Giovannoni G, Southam E, Waubant E. Systematic review of disease-modifying therapies to assess unmet needs in multiple sclerosis: tolerability and adherence. Mult Scler 2012 Jul;18(7):932-46.
- Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006;354:899–910.
- National Institute for Health and Care Excellence. Final appraisal determination—Fingolimod for the treatment of highly active relapsing–remitting multiple sclerosis. 2012.
- Brinkmann V, Davis MD, Heise CE et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem 2002;277(24):21453–21457.
- Matloubian M, Lo CG, Cinamon G et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004;427(6972):355–360.
- Choi JW, Gardell SE, Herr DR et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci USA 2011;108(2):751–756.
- Kovarik JM, Schmouder R, Barilla D, et al. Multiple-dose FTY720: tolerability, pharmacokinetics, and lymphocyte responses in healthy subjects. J Clin Pharmacol 2004;44(5),532–537.
- Schmouder RL, Choudhury S, Barilla D, et al. Prolonged, consistent oral absorption of FTY720. Am J Transplant 2001;1:475.
- Richard NR, Giannetti P, Alsanousi A, et al. Development of oral immunomodulatory agents in the management of multiple sclerosis. Drug Des Devel Ther 2011;5:255-74.
- Kappos L, Antel J, Comi G et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med 2006;355(11):1124–1140.
- Calabresi PA. Efficacy and safety of fingolimod in patients with relapsing–remitting multiple sclerosis (RRMS): results from an additional 24-month double-blind, placebo-controlled study (FREEDOMS II Study). Presented at: American Academy of Neurology 64th Annual Meeting. New Orleans, LA, USA, 25 April 2012.
- Khatri B, Barkhof F, Comi G et al.; TRANSFORMS Study Group. Comparison of fingolimod with interferon β-1a in relapsing–remitting multiple sclerosis: a randomised extension of the TRANSFORMS study. Lancet Neurol 2011;10(6):520–529.
- Singer BA. Fingolimod for the treatment of relapsing multiple sclerosis. Expert Rev Neurother 2013;13(6):589-602.
- Cohen JA, O’Connor P, Caliolio T et al. Long-term safety of fingolimod in relapsing multiple sclerosis: update to integrated analyses of Phase 2 and 3 studies and extension phases. Presented at: 28th Congress of the European Committee for Treatment and Research in Multiple Sclerosis. Lyon, France, 10–13 October 2012.
- Guo J, MacDonell KL, Giles, WR. Effects of sphingosine 1-phosphate on pacemaker activity in rabbit sino-atrial node cells. Pflugers Arch 1999;438(5):642–648.
- Francis G, Kappos L, O’Connor P, et al. Lymphocytes and fingolimod: temporal pattern and relationship with infections. Presented at: American Academy of Neurology Annual Meeting. Honolulu, HI, USA, 9-16 April 2011.
- Brinkmann V, Cyster JG, Hla T. FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am J Transplant 2004;4(7):1019-25.
- GilenyaTM. Summary of product charcteristics. Novartis Europharm Limited, UK (2011).
- Geissbuhler Y, Butzkueven H, Hernandez-Diaz S, et al. Pregnancy outcomes from fingolimod clinical trials and post-marketing experience and the need for a multinational GilenyaTM (fingolimod) Pregnancy Exposure Registry in Multiple Sclerosis. Presented at: 28th Congress of the European Committee for Treatment and Research in Mutlitple Sclerosis. Lyon, France, 12 October 2012.