Biohaven achieves positive results in pivotal study
September 23 2024
- Troriluzole 200 mg dosed orally, once daily, in patients with spinocereballar ataxia (SCA) met the study’s primary endpoint on the change from baseline in the modified functional Scale for the Assessment and Rating of Ataxia (f-SARA) at 3 years in all study population genotypes.
- Troriluzole also showed statistically significant superiority after both 1 and 2 years of treatment.
- Troriluzole achieved statistically significant superiority on 9 consecutive, prespecified primary and secondary endpoints.
- SCA patients treated with troriluzole showed a 50-70% slowing of disease progression, representing 1.5-2.2 years delay in disease progression over the 3-year study period.
- Biohaven plans to submit a New Drug Application (NDA) to the US Food and Drug Administration (FDA) for troriluzole in the treatment of all spinocereballar ataxia (SCA) genotypes in 4Q 2024. The application is eligible for a priority review given orphan drug and fast-track designations previously granted by FDA.
Biohaven Ltd have announced positive topline results from pivotal Study BHV4157-206-RWE (NCT06529146) demonstrating the efficacy of troriluzole on the mean change from baseline in the f-SARA after 3 years of treatment. The study achieved the primary endpoint and showed statistically significant improvements on the f-SARA at years 1 and 2 (Figure 1). SCA is a rare, progressively debilitating neurodegenerative disease that affects approximately 15,000 people in the United States and 24,000 in Europe and the UK. There are no FDA approved treatments for SCA.
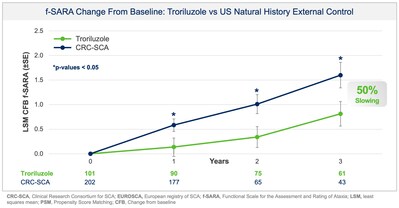
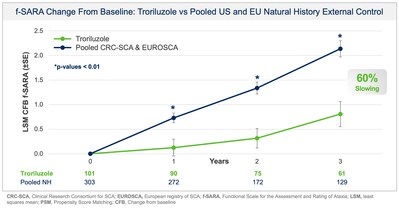
Collectively, data across multiple analyses demonstrate a robust and clinically meaningful slowing of disease progression in SCA patients. These treatment benefits translate into a 50-70% slower rate of decline compared to untreated patients, representing 1.5-2.2 years delay in disease progression over the 3-year study period. Additionally, in a responder sensitivity analysis, disease progression when defined by a 2 point or greater worsening on the f-SARA at 3 years showed an odds ratio (OR) of 4.1 (95% CI: 2.1, 8.1) for the untreated external control arm versus troriluzole treated subjects (p < 0.0001; pooled analysis).
Dr. Susan Perlman, Director of Ataxia Clinic and Neurogenetics Clinical Trials at the David Geffen School of Medicine at UCLA stated, “SCA is a debilitating, relentlessly progressive disease that destroys quality of life, leaving patients unable to care for themselves, walk, or speak. Troriluzole is the very first treatment to show a delay in disease progression that can give patients additional years of independence, where they can walk without assistance, continue to work, play with their children, and participate in daily activities. This is an exciting and hopeful moment for the SCA community.”
Study BHV4157-206-RWE was designed, in discussion with the US Food and Drug Administration (FDA), to assess the effectiveness of troriluzole in SCA after 3 years of treatment as measured by the change from baseline in the f-SARA. The study utilised Phase 3 data and an external control of matched, untreated SCA subjects from the US Clinical Research Consortium for the Study of Cerebellar Ataxia (CRC-SCA) in accordance with FDA’s Guidance on Real-World Evidence (RWE) of effectiveness. All endpoints were prespecified, and both the study protocol and statistical analysis plan were submitted to, and reviewed by, FDA prior to topline data analysis. The new analysis doubled the previously available 3 year data with 63 subjects now completing 3 years of treatment with troriluzole and matched to the external control arm. Propensity Score Matching (PSM) was used to ensure that untreated patients from the CRC-SCA study were rigorously matched to treated patients from Study BHV4157-206 on baseline characteristics. The primary objective was to examine the treatment effects of troriluzole for up to 3 years, by comparing data on the f-SARA from patients treated with troriluzole in Study BHV4157-206 to untreated patients from the natural history study. Troriluzole-treated patients demonstrated statistically significant and sustained benefits at years 1, 2 and 3 on the f-SARA compared to a rigorously matched natural history control.
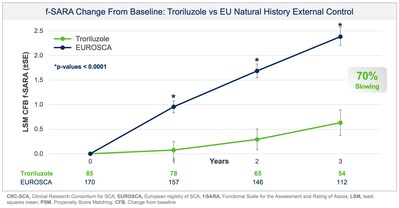
Additionally, prespecified analyses in the protocol employed a separate, independent natural history control from the European SCA natural history study (EUROSCA) for global regulatory purposes. Results using the EUROSCA patients, in addition to a pooled analysis using both CRC-SCA and EUROSCA patients, as the external controls were also statistically significant and consistent with the primary efficacy analysis at all timepoints (see Figure 2 and Figure 3). The addition of EUROSCA data increased the external control sample size and added to the robustness of the statistically significant treatment differences at years 1, 2, and 3, favoring troriluzole.
Jeremy Schmahmann, M.D., Professor of Neurology at Harvard Medical School and Founding Director of the Ataxia Center at Massachusetts General Hospital commented:
The stabilisation of SCA symptoms as reflected by the topline data at 3 years along with the previously reported reductions in falls show the therapeutic potential of troriluzole. I cannot underscore enough the impact of a potential treatment that can slow SCA disease progression and the effect on patients and caregivers who have helplessly watched generations of family members deteriorate and die from SCA. These new data provide support for troriluzole as a safe and effective once daily treatment for patients with SCA.
Spinocerebellar ataxia is a group of dominantly inherited neurodegenerative disorders characterised by progressive loss of voluntary motor control and atrophy of the cerebellum, brainstem and spinal cord. Patients experience significant morbidity, including progression to a wheelchair, impaired gait leading to falls, inability to communicate due to speech impairment, difficulty swallowing, and premature death. While signs and symptoms can appear anytime from childhood to late adulthood, SCA typically presents in early adulthood and progresses over a number of years. Currently, there are no FDA-approved treatments and no cure for SCA.
Based upon the topline data from Study BHV4157-206-RWE, and previous safety and efficacy data from the troriluzole development programme in SCA, Biohaven plans to submit a New Drug Application (NDA) to the FDA in Q4 2024. The troriluzole development programme has generated the largest clinical trial dataset in SCA and now has follow-up in some patients treated with troriluzole for over 5 years. Biohaven has previously received both Fast-Track and Orphan drug designation (ODD) from the FDA, and ODD from the European Medicines Agency, for troriluzole in SCA. An NDA with ODD is eligible for priority FDA review. Biohaven will be prepared to commercialise SCA in the US in 2025, if ultimately approved, based on potential priority review timelines.