LONDON (February 13, 2020) The Cure Parkinson’s Trust (CPT) and Van Andel Institute (VAI) have announced a new three-year co-funding agreement that pledges USD $4.5 million to Parkinson’s research, strengthening an already long-standing partnership.
This funding will support the International Linked Clinical Trials (iLCT) initiative, a thriving global programme that aims to develop new, potentially disease-modifying Parkinson’s therapies, many of which are repurposed medications originally designed or approved to treat other diseases. Candidate drugs are evaluated and prioritised annually by a committee of world-leading Parkinson’s experts, who select which drugs should enter clinical trials in people with Parkinson’s. These drugs have the potential to get to the clinic much faster as they have already passed crucial safety tests.
Currently, 15 trials of drugs evaluated by the committee are underway and seven trials have been completed. In addition, a further 10 trials are in the planning stages. To date, iLCT has included more than 2500 trial participants.
Since its inception in 2012, CPT’s and VAI’s involvement has ensured that each year the iLCT initiative goes from strength to strength as progressively more clinical trials of potentially disease-modifying drugs are launched within the programme.
The iLCT initiative has already shown positive results, most recently in the phase II clinical trial of ambroxol led by Professor Anthony Schapira of the UCL Queen Square Institute of Neurology. Ambroxol is a drug prioritised by the iLCT committee in 2014 and currently in use to treat respiratory conditions. The results, published in The Journal of the American Medical Association in January, revealed that ambroxol was safe and well tolerated by participants. Importantly, the results also showed the drug was able to cross the blood brain barrier and increase levels of glucocerebrosidase (GCase) in the brain cells of people with Parkinson’s. The protein GCase allows cells to remove waste more effectively, a function which evidence suggests is deficient in some people with Parkinson’s. Increasing levels of this protein may have the potential to keep cells healthier for longer and, therefore, slow Parkinson’s progression.
In addition to the new funding for the iLCT programme, VAI and CPT together with the John Black Charitable Foundation have agreed to co-fund £522,000 (or USD $679,000) of the next phase in a programme of research into ambroxol to assist the drug to move forward into phase III trials. The first step, led by Professor Schapira of UCL, is to determine the optimal dose of the drug.
Professor Patrik Brundin, Chair of the Linked Clinical Trials committee and Director of the Center for Neurodegenerative Science at VAI said:
We are thrilled to continue our long-standing collaboration with The Cure Parkinson’s Trust on the International Linked Clinical Trials initiative and look forward to expanding our program to evaluate additional promising medications in the coming years. We are especially grateful to the trial participants, without whom this critical work would not be possible. I am immensely hopeful that, together, we will find a way to slow or stop Parkinson’s progression.
Will Cook, CEO of CPT said
CPT is delighted to announce this funding agreement, which builds on the ground-breaking relationship the charity has developed with VAI since 2012. This will enable the launch of many more clinical trials of potentially disease-modifying repurposed and novel drugs that have been identified through the diligent iLCT process, and thereby bringing us closer to our goal: a cure for the 10 million people living with Parkinson’s globally.
Dr Richard Wyse, Director of Research & Development at CPT said
The massive clinical undertaking involving so many drugs repurposed from other therapeutic areas is unique not only in neurology, but is larger than any drug repositioning programme, whether academic or commercial, in any other disease. The partnership between VAI and CPT bears testament to the principle of collaboration and we look forward to continuing and growing this partnership in the years ahead.
– Statistically significant results in primary and key secondary endpoints
– Risdiplam is the first SMA treatment to have positive placebo-controlled data in pivotal studies across a real world population of infants, children, teenagers and adults-
SOUTH PLAINFIELD, N.J., February 6, 2020 PTC Therapeutics, Inc. today announced that positive 1 year clinical data from Part 2 of the pivotal SUNFISH study evaluating risdiplam in people aged 2-25 years with nonambulatory type 2 and type 3 spinal muscular atrophy (SMA) were presented at the 2nd International SMA Europe Conference.
The study demonstrated that the change from baseline in the primary endpoint Motor Function Measure scale (MFM-32) was significantly greater in patients receiving risdiplam compared to placebo (1.55 point mean difference; p=0.0156) at 12 months of treatment. As expected, the age-related effect was notable and consistent with SMA disease progression, with younger patients (2-5 years) (78.1% vs 52.9% achieving ≥3 point increase) showing clear improvement relative to placebo while older patients demonstrated disease stabilization and, in some cases, improvement. Safety for risdiplam was consistent with its known safety profile and no new safety signals were identified.
“The results of this ambitious study confirm risdiplam’s potential to be the most competitive global product for a broad range of SMA patients,” said Stuart W. Peltz, Ph.D., Chief Executive Officer of PTC Therapeutics.
Current approved treatments only address a portion of SMA patients. This was the first clinical study encompassing SMA patients representative of the population that are seen in clinical practice. Together with the positive data from the FIREFISH study in type 1 patients, these data exemplify the potential of risdiplam for all SMA patients.
SUNFISH is a two-part, double-blind, placebo-controlled, pivotal study in people aged 2-25 years (n=180) with type 2 and 3 SMA, with broad inclusion criteria. The majority of patients in the study were older, had more progressed disease as evidenced by severe scoliosis and contractures, and had lower baseline scores on motor function scales relative to other clinical trials in this patient population. The primary endpoint for Part 2 was change at 12 months in motor function as measured by the MFM-32 scale compared to placebo. The MFM-32 scale is a highly sensitive, validated measure used to evaluate fine and gross motor function and is a more relevant measure for patients who have more progressed disease.
The pivotal, Part 2 component of the study met its primary endpoint (cut-off date: Sep 6, 2019). Total mean change from baseline in the MFM-32 score was significantly greater in patients receiving risdiplam compared to those treated with placebo at 12 months (p=0.0156).
The strongest responses in MFM-32 compared to placebo were observed in the youngest age group (2-5 years) (78.1% vs 52.9% achieving ≥3 point increase) as expected.
Stabilization, which is the goal of treatment in the older age group (18-25 years) with more established disease, was achieved versus placebo (57.1% vs 37.5%, with stabilisation defined as a ≥0 point increase).
Medically meaningful and statistically significant results were demonstrated in key secondary endpoints.
Revised Upper Limb Module (RULM) mean change from baseline was significantly greater in patients receiving risdiplam compared with placebo (1.59 point difference; p=0.0028).
In the Hammersmith Functional Motor Scale Expanded (HFMSE) there was a numerical difference in favour of risdiplam which did not reach statistical significance (p=0.3015).
Patients and caregivers also reported numerical improvements in independence as measured by the SMA Independence Scale, a new measurement that captures highly relevant, day-to-day activities such as eating and drinking, getting dressed, and overall hygiene, amongst many other daily activities. Patients and caregivers reported improvements in independence after treatment with risdiplam over 12 months (Caregiver (for all patients), p=0.0022; Patients (≥12 years), p=0.1778).
More than 400 patients have been treated with risdiplam across all studies to date, with no treatment-related safety findings leading to study withdrawal in any risdiplam trial. The adverse event profile was similar to placebo. The most common adverse events were upper respiratory tract infection (31.7%), nasopharyngitis (25.8%), pyrexia (20.8%), headache (20%), diarrhea (16.7%), vomiting (14.2%) and cough (14.2%). While the rate of lower respiratory tract infections overall was similar in both treatment arms (RIS 19%, PLB 20 %), serious lower respiratory tract infections occurred in more patients in the risdiplam group (RIS 10%, PLB 2%) but were reported as unrelated to risdiplam and resolved without change to study treatment.
Risdiplam (RG7916), is an investigational, oral, first-in-class, mRNA splicing modifier for the treatment of SMA. Recently, positive results from the pivotal single-arm FIREFISH Part 2 study, which assessed the efficacy of risdiplam in 41 infants (eligible age at enrollment between 1 and 7 months) with type 1 SMA treated for 12 months, were announced. The SMA programme is a collaboration between PTC, the SMA Foundation, and Roche.
In November 2019, the U.S. Food and Drug Administration (FDA) accepted a New Drug Application (NDA) and granted Priority Review for risdiplam. The NDA filing was based on 12-month data from the dose-finding, Part 1 of the pivotal FIREFISH and SUNFISH studies, and preclinical pharmacokinetic and clinical and pharmacodynamic data in all types of SMA. The Prescription Drug User Fee Act (PDUFA) goal date for a decision by the FDA is May 24, 2020.
About Spinal Muscular Atrophy (SMA)
Spinal muscular atrophy (SMA) is a severe, inherited, progressive neuromuscular disease that causes devastating muscle atrophy and disease-related complications. It is the most common genetic cause of infant mortality and one of the most common rare diseases, affecting approximately one in 11,000 babies. SMA leads to the progressive loss of nerve cells in the spinal cord that control muscle movement. Depending on the type of SMA, an individual’s physical strength and their ability to walk, eat or breathe can be significantly diminished or lost.
SMA is caused by a mutation in the survival motor neuron 1 (SMN1) gene that results in a deficiency of SMN protein. SMN protein is found throughout the body and increasing evidence suggests SMA is a multi-system disorder and the loss of SMN protein may affect many tissues and cells, which can stop the body from functioning.
About risdiplam
Risdiplam is an investigational survival motor neuron2 (SMN2) splicing modifier for SMA and is an orally administered liquid. It is designed to durably increase and sustain SMN protein levels both throughout the central nervous system and in peripheral tissues of the body. Risdiplam is being studied in a broad clinical trial programme in SMA, with patients ranging from birth to 60 years old, and includes patients previously treated with other SMA-targeting therapies. The clinical trial population represents the broad, real-world spectrum of people living with this disease. The risdiplam clinical development programme was designed with the aim of enabling access for all appropriate patients.
Risdiplam is currently being evaluated in four multicentre trials in people with SMA:
SUNFISH (NCT02908685) – SUNFISH is a two-part, double-blind, placebo-controlled pivotal study in people aged 2-25 years with types 2 or 3 SMA. Part 1 (n=51) determined the dose for the confirmatory Part 2. Part 2 (n=180) evaluated motor function using total score of Motor Function Measure 32 (MFM-32) at 12 months. MFM-32 is a validated scale used to evaluate fine and gross motor function in people with neurological disorders, including SMA.
FIREFISH (NCT02913482) – an open-label, two-part pivotal clinical trial in infants with type 1 SMA. Part 1 was a dose-escalation study in 21 infants. The primary objective of Part 1 was to assess the safety profile of risdiplam in infants and determine the dose for Part 2. Part 2 is a pivotal, single-arm study of risdiplam in 41 infants with type 1 SMA treated for 24 months, followed by an open-label extension. Enrollment for Part 2 was completed in November 2018. The primary objective of Part 2 is to assess efficacy as measured by the proportion of infants sitting without support after 12 months of treatment, as assessed in the Gross Motor Scale of the Bayley Scales of Infant and Toddler Development – Third Edition (BSID-III) (defined as sitting without support for 5 seconds).
JEWELFISH (NCT03032172) – an open-label exploratory trial in people with SMA aged 6 months–60 years who have been previously treated with SMA-directed therapies. The study has completed recruitment.
RAINBOWFISH (NCT03779334) – an open-label, single-arm, multicentre study, investigating the efficacy, safety, pharmacokinetics and pharmacodynamics of risdiplam in babies (~n=25), from birth to six weeks of age (at first dose) with genetically diagnosed SMA who are not yet presenting with symptoms. The study is currently recruiting.
About PTC Therapeutics, Inc.
PTC is a science-driven, global biopharmaceutical company focused on the discovery, development and commercialization of clinically differentiated medicines that provide benefits to patients with rare disorders. PTC’s ability to globally commercialize products is the foundation that drives investment in a robust and diversified pipeline of transformative medicines and our mission to provide access to best-in-class treatments for patients who have an unmet medical need.
Emergency hospital admissions for people with multiple sclerosis (MS) are still on the rise and costing the NHS millions; yet many of them could be avoided, according to a new summary of data by healthcare intelligence provider Wilmington Healthcare and educator MS Academy.
The information, which updates the authors’ previous annual analysis of Hospital Episode Statistics (HES) from 2016/17, shows that emergency hospital admissions for people with MS in England have continually increased over the past three years. During 2018/19, 17.3% of people living with MS were admitted for an emergency, an increase of over 2,000 people.
According to the latest HES data, there were 30,310 emergency hospital admissions for people with MS in England during this time with a cost to the NHS of £86 million. This is an increase of £16 million when compared to 2016/17 data.
The average cost per emergency admission was £2,844 and the average length of stay was 7.6 days – yet a large proportion of this care could have been prevented.
Many of the emergency admissions were for problems that can be avoided with proactive, preventative care, earlier diagnosis and intervention in the community. For example, the report found that emergency admissions for respiratory issues like infections cost the NHS £15 million, whilst bladder and bowel problems cost £9.1 million.
Gavin Giovannoni, Professor of Neurology for the Blizard Institute, Barts and the London School of Medicine and Dentistry and Academic Director for MS Academy said:
We thought we were making progress, or at least treading water when it came to managing the complications of MS. However, the latest NHS England data suggests we are going backwards. It is clear that unplanned emergency admissions related to MS are increasing in number and cost to the NHS. In addition, the profile of complications is changing with admissions for respiratory issues topping the list. Does this mean that we have successfully tackled urinary tract infections? Unlikely as they continue to be a massive problem.
Can we do more with less? We think we can and this is why it is important for the wider MS community to engage with our activities in relation to raising the bar and tackling variation in the provision of MS services across the country. Without working differently and ensuring that no patient with MS is left behind we are not going to reverse these trends.
Sue Thomas, CEO of Wilmington Healthcare Consulting, said:
“Our latest report shows that the problems we have highlighted for people with MS in our annual analysis from as far back as 2013/14 only continue to grow.
“This is one reason why additional MS nurses, allied health professionals and neuro pharmacists are needed in the UK to provide regular assessment and preventative management strategies. With the right specialists and community-based support the NHS could significantly reduce the need for emergency hospital care for people with MS.”
“Preventative care strategies for people with MS must be reviewed in order to tackle problems, such as respiratory and urinary tract infections, at an early stage to try to avert the need for emergency care. This would not only be of huge benefit to patients, it would also reduce pressure on struggling A&E departments.”
MS Academy is working in partnership with voluntary sector and patient organisations and healthcare professionals to highlight the variation in current service provision. Managing Director Sarah Gillett said:
“It is disappointing to see that avoidable emergency hospital admissions for people with MS have continued to rise in the past year.
“Over the last two years, MS Academy has brought together partners from across the MS sector to highlight how current services need to change and to work practically together to deliver that change. Our ‘Raising the Bar’ work this year will continue to drive forward this agenda to address unwarranted variation and improve the experiences of people affected by MS.”
STRIDE registry analysis shows Translarna preserved ambulation and physical function by years compared with those in CINRG Duchenne Natural History Study, with no new safety signals1 – Trend toward delayed worsening of pulmonary function compared with natural history study1
SOUTH PLAINFIELD, NJ., February 3, 2020 – PTC Therapeutics, Inc. (NASDAQ:PTCT) announced real-world data showing that boys with nonsense mutation Duchenne muscular dystrophy treated with Translarna (ataluren) and standard of care (SoC), preserved the ability to walk for years longer than those on SoC alone. Pulmonary function was also preserved in those treated with Translarna.1 The analysis, presented in the publication of an interim analysis of preliminary real-world data, compared children treated with Translarna in a real-world setting from the STRIDE[1] registry with a matched cohort in a long-term natural history study, CINRG[2].1 In addition, no new safety signals were observed in the patients treated with Translarna, consistent with what has been shown in previous clinical trials.1 The interim data have been published in the Journal for Comparative Effectiveness Research. Final data from the STRIDE registry is expected in 2025.
“Duchenne muscular dystrophy is a devastating disease that causes irreversible muscle wasting and progressively robs young people of their ability to walk, move, and breathe naturally without a ventilator, and it reduces their autonomy in daily life tasks,” said Dr. Andrés Nascimento, Pediatric Neurology, Neuromuscular Diseases Unit, SJD Children’s Hospital, Barcelona, Spain.
In a real-world setting, children and adolescents treated with Translarna experience a delay in the disease progression, are able to maintain more mobility, and have a higher level of physical autonomy concerning the course of the natural history of the disease. This is not only clinically relevant, but especially important for the quality of life of patients and their families.
Children treated with Translarna in a real-world setting as part of the STRIDE registry were able to walk independently for an additional 3.5 years compared with a propensity-score matched cohort in the CINRG natural history study, with a median age at loss of ambulation of 14.5 years and 11 years, respectively (72% relative risk reduction).1
Additional analyses from the registry demonstrated that Translarna sustained the ability of boys with Duchenne to complete everyday tasks by years compared with the natural history cohort.1 In timed function tests, Translarna sustained their ability to stand up from lying down, in under 5 and 10 seconds, for three years longer than in boys treated with SoC alone.1 Boys treated with Translarna were also still able to climb four stairs in under 5 and 10 seconds for 1.5 and 3.6 years longer, respectively, than boys on SoC alone.1
In addition, the analysis showed a trend toward delayed worsening of pulmonary function in routine clinical practice for patients treated with Translarna, compared to the matched patients in CINRG, Researchers evaluated FVC, a traditional measure of lung function in Duchenne patients that correlates with disease progression and mortality.1,2 The STRIDE data showed that 32.1% of standard of care patients from the natural history cohort had an FVC of <50%, compared to only 2.2% of patients receiving Translarna.1,2 However, the authors state that given the low number of events and the shorter duration of follow-up of patients in the STRIDE registry compared to CINRG in these interim analyses, it is premature to draw firm conclusions from these results.1 After loss of ambulation and loss of the use of the arms, the respiratory muscles of people with Duchenne start to progressively deteriorate, leading to the risk of life-threatening respiratory complications and the need for ventilation support.1
“The data from the STRIDE registry are consistently confirming the benefits seen in Translarna clinical trials and the difference it is making to patients and their families – more years of being independent and physically able without reliance on a wheelchair or ventilator,” said Dr. Claudio Santos, SVP, Global Medical Affairs, PTC Therapeutics.
About Translarna (ataluren)
Translarna (ataluren), discovered and developed by PTC Therapeutics, Inc., is a protein restoration therapy designed to enable the formation of a functioning protein in patients with genetic disorders caused by a nonsense mutation. A nonsense mutation is an alteration in the genetic code that prematurely halts the synthesis of an essential protein. The resulting disorder is determined by which protein cannot be expressed in its entirety and is no longer functional, such as dystrophin in Duchenne muscular dystrophy. Translarna, the tradename of ataluren, is licensed in the European Economic Area for the treatment of nonsense mutation Duchenne muscular dystrophy in ambulatory patients aged two years and older. Ataluren is an investigational new drug in the United States.
About the STRIDE Registry
The STRIDE (Strategic Targeting of Registries and International Database of Excellence) Registry is an ongoing, multicentre, observational study of the safety and effectiveness of Translarna in routine care. It is the first patient data repository to provide real-world experience regarding the long-term use of Translarna in routine clinical practice. Enrolled patients will be followed for at least 5 years from the date of enrolment, or until withdrawal from the study.As of 9 July 2018, 217 patients with a mean age of 9.8 years had been enrolled across 11 countries in Europe and Israel.
Effectiveness information may include neuromuscular function (as measured for example by timed-function tests, the North Star Ambulatory Assessment, and Performance of the Upper Limb (PUL) measures, cardiac function (including echocardiogram where available), pulmonary function (including spirometry measures), and quality of life measures. Assessments of musculoskeletal health, rehabilitation, orthopedic and gastrointestinal management, as well as other measures of psychosocial management, will be collected to allow for comparison of patient health-management activities in routine clinical care to those of published treatment guidelines.
STRIDE is a collaborative partnership between TREAT-NMD and PTC Therapeutics, led by a Steering Committee comprised of leading experts in Duchenne, patient advocates from around the world and PTC representatives.
The Registry also fulfils a post-marketing commitment to the Pharmacovigilance Risk Assessment Committee of the European Medicines Agency.
About TREAT-NMD
TREAT-NMD is a network for the neuromuscular field that provides an infrastructure to ensure that the most promising new therapies reach patients as quickly as possible. Since its launch in January 2007 the network’s focus has been on the development of tools that industry, clinicians and scientists need to bring novel therapeutic approaches through preclinical development and into the clinic, and on establishing best-practice care for neuromuscular patients worldwide. The network has developed from its European roots to become a global organization that brings together leading specialists, patient groups and industry representatives to ensure preparedness for the trials and therapies of the future while promoting best practice today.
The Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study (DNHS; ClinicalTrials.gov identifier: NCT00468832) was a prospective, longitudinal study of more than 400 patients with Duchenne muscular dystrophy (DMD) who were followed up between 2006 and 2016 at 20 worldwide centers as part of the academic clinical trial network, CINRG.
The CINRG DNHS enrolled patients aged 2–28 years with DMD at 20 centers in nine countries from 2006 to 2016. Data from CINRG DNHS patients receiving standard of care (SoC) are used in the present analysis as a control to provide context for assessing the effects of ataluren plus SoC in patients in the STRIDE Registry. SoC refers to palliative therapies and corticosteroid treatment.
About Duchenne Muscular Dystrophy
Primarily affecting males, Duchenne muscular dystrophy (Duchenne) is a rare and fatal genetic disorder that results in progressive muscle weakness from early childhood and leads to premature death in the mid-twenties due to heart and respiratory failure. It is a progressive muscle disorder caused by the lack of functional dystrophin protein. Dystrophin is critical to the structural stability of all muscles, including skeletal, diaphragm, and heart muscles. Patients with Duchenne can lose the ability to walk as early as age ten, followed by loss of the use of their arms. Duchenne patients subsequently experience life-threatening lung complications, requiring the need for ventilation support, and heart complications in their late teens and twenties.
More information regarding Duchenne is available through the Muscular Dystrophy Association and the Parent Project Muscular Dystrophy. Additionally, information and resources are available at http://www.duchenneandyou.com
About PTC Therapeutics, Inc.
PTC is a science-driven, global biopharmaceutical company focused on the discovery, development and commercialization of clinically-differentiated medicines that provide benefits to patients with rare disorders. PTC’s ability to globally commercialize products is the foundation that drives investment in a robust pipeline of transformative medicines and our mission to provide access to best-in-class treatments for patients who have an unmet medical need. To learn more about PTC, please visit us on http://www.ptcbio.com and follow us on Facebook, on Twitter at @PTCBio, and on LinkedIn.
– Study meets primary endpoint in patients living with type 1 SMA
– To date, more than 400 patients have been treated with risdiplam across all studies, with no treatment related safety findings leading to study withdrawal in any risdiplam trial
– Risdiplam PDUFA expected May 24, 2020
SOUTH PLAINFIELD, N.J., January 23, 2020 – PTC Therapeutics, Inc. today announced positive topline results from part 2 of FIREFISH demonstrating that the study met its primary endpoint of proportion of infants who are sitting without support after 12 months of treatment. The pivotal study assessed the efficacy of risdiplam (RG7916) in infants with type 1 spinal muscular atrophy (SMA), the most severe, infantile onset form of this rare and devastating neuromuscular disease. Risdiplam has been well tolerated and no treatment-related safety findings leading to withdrawal have been observed in any risdiplam trial to date. Data from part 2 of the FIREFISH study will be shared with health authorities globally and will be presented at an upcoming medical congress.
“We are excited about the FIREFISH results as they demonstrate how effective risdiplam is in type 1 SMA patients, where developmental milestones such as rolling, sitting and standing were achieved,” said Stuart W. Peltz, Ph.D., Chief Executive Officer of PTC Therapeutics. “These results further support the growing body of evidence of risdiplam’s benefit in SMA patients across all types studied and reinforce the potential of our small molecule splicing platform to identify new therapies for patients who have limited or no treatment options.”
The single-arm part 2 of FIREFISH study assessed the efficacy of risdiplam in 41 infants (eligible age at enrollment between 1 and 7 months) with type 1 SMA treated for 12 months. The primary endpoint in the study was defined as proportion of infants who are sitting without support after 12 months of treatment as assessed by the Bayley gross motor scale of infant and toddler development – third edition (BSID-III). Safety for risdiplam was consistent with its safety profile to date and no new safety signals were identified. No treatment-related safety findings leading to study withdrawal have been observed.
Risdiplam (RG7916), is an investigational, potential first-in-class oral mRNA splicing modifier for the treatment of SMA. FIREFISH is an open-label, two-part pivotal clinical trial in infants with type 1 SMA. Part 1 was a dose-finding study in 21 infants. The primary objectives of part 1 were to evaluate the safety profile of risdiplam in infants and to determine the dose for part 2. Part 1 of the trial showed that infants with type 1 SMA survived and achieved developmental milestones beyond those expected in the natural course of the disease. The SMA program is a collaboration between PTC, the SMA Foundation, and Roche.
Recently, positive results from part 2 of SUNFISH, a study evaluating the efficacy and safety of risdiplam in patients between 2 and 25 years of age with type 2 or 3 SMA, were announced. In November 2019, the U.S. Food and Drug Administration (FDA) accepted a New Drug Application (NDA) and granted Priority Review for risdiplam. The NDA filing was based on 12-month data from the dose-finding part 1 of the pivotal FIREFISH and SUNFISH studies, and preclinical pharmacokinetic and clinical and pharmacodynamic data in all types of SMA. The Prescription Drug User Fee Act (PDUFA) goal date for a decision by FDA is May 24, 2020.
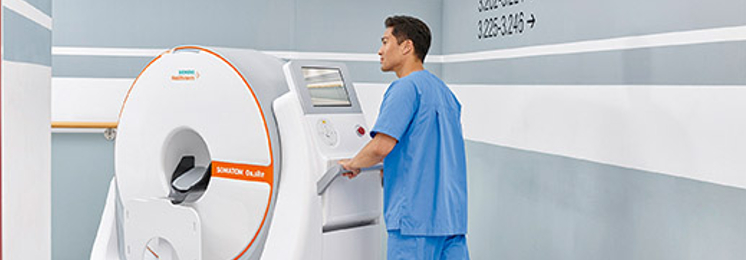
Transporting ICU patients with acute and critical head conditions to the radiology department for CT imaging is staff- and time-intensive. Time that could otherwise be spent on patient care. Siemens have developed SOMATOM On.site, a new mobile head CT scanner that allows scanning of patients directly on the ICU ward.
Neurokinex Kids – a paediatric spinal cord injury rehabilitation facility near Gatwick – is bringing breakthrough therapies to children living with Cerebral Palsy. The success of its protocols and approach comes not a moment too soon with the number of children with CP in England and Wales estimated to increase 7.5% percent by 20201.
Neurokinex Kids’ therapies for Cerebral Palsy focus primarily on relieving spasticity and improving strength and range of motion. Emphasis is put on developing motor control and functional skills, including gait, balance and transfers in and out of a wheelchair. Fine motor skills and bilateral hand coordination are also worked on with the overall aim to increase children’s independence in everyday activities.
Neurokinex delivers specialist neurological rehabilitation and therapies tailored to each child’s needs and typically look to address:
Increase range of motion in the lower body (hips, legs, ankles, and feet) and upper body (shoulder, elbow, wrists and hands)
Increase strength and motor control
Improve balance and coordination
Develop and improve walking and transitional movements
Develop and improve gross and fine motor skills
Incorporate new patterns of moving into functional skills
Develop smooth coordinated movement
In most CP cases, the neural pathways develop intact prior to birth so have the capacity to function. Our programmes set out to excite the nervous system and awaken dormant pathways while also treating the life-impacting symptoms presented by the disability. We work differently to traditional rehab or physiotherapy by using activity-based rehabilitation (ABR) techniques that involve the whole body in activity
Jenny Suggitt, Occupational Therapist and Centre Manager at Neurokinex Gatwick
At Neurokinex Kids, children leave their wheelchairs and take part in assisted standing, stepping, climbing and playing. Not only does this optimise the strength and endurance of the muscles that are functioning, it also stimulates the muscles affected by the condition. Neurokinex tailors its therapies to each child to suit their ability and age and is treating upwards of 30 children aged two to 14 years old. The therapies are presented as play with the Neurokinex Kids gym providing the perfect backdrop to put the ‘fun’ back into functional movement.
“Cerebral Palsy is a neurological condition and, although there is limited research specific to children with CP, we are seeing success through applying research completed on adults with neurological conditions,” explains Jenny. “Overall, we try to generate reflex activity in order to allow them to gain experience of the movements,” she says. “By physically assisting them into positions that allow their body to experience these sensations, we hope they can gain voluntary control over those movements.”
Two key protocols
Neurokinex is an affiliated partner of the NeuroRecovery Network, established by the late Superman actor Christopher Reeve and his wife Dana. The only affiliate outside the US, Neurokinex is able to use its two unique protocols to achieve success.
Locomotor Training (LT) comprises a sophisticated treadmill programme that aims to reawaken dormant nerve pathways by repetitively stimulating nerves and muscles in the lower body to retrain the spinal cord to ‘remember’ the pattern of walking. LT improves trunk control in children with recent research (1) showing 100% of children improved their trunk control after the completion of intensive LT blocks (1.5h per day, 5 days per week, for 60 sessions)
Wide Pulse Stimulation (WPS) targets many muscles at the same time when administered as part of active therapy. Applying an electrical stimulus through electrodes as the individual performs an exercise or functional task signals the central nervous system to develop new pathways or strengthen existing ones.
On 20th January 2020, Novartis announced that Mayzent® (siponimod) has been approved as the first and only oral treatment specifically indicated for patients with secondary progressive multiple sclerosis (SPMS) with active disease in Europe.1
Mayzent addresses an unmet need for SPMS patients with active disease who, until now, did not have an oral treatment that has been shown to be effective in delaying progression in this patient population.
Approval is based on the Phase III EXPAND trial, the largest randomised clinical study in a broad range of SPMS patients, showing Mayzent significantly reduced the risk of disease progression, including physical disability and cognitive decline2,3
Kappos L, Cree B, Fox R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomized, phase 3 study. Lancet. Published online March 22, 2018. http://dx.doi.org/10.1016/S0140-6736(18)30475-6.
Mayzent® (siponimod) Summary of Product Characteristics. Novartis International AG. January 2020.
Results of a Phase II clinical trial evaluating ambroxol as a potential treatment to slow progression of Parkinson’s (PD) have shown it can effectively cross the blood-brain barrier and increase levels of glucocerebrosidase (GCase) in the brain cells of people with PD. Between January 2017 and April 2018 a Phase II clinical trial was conducted by Professor Anthony Schapira and his research team at University College, London and the Royal Free Hospital. Results were published in The Journal of the American Medical Association (JAMA) Neurology on 13th January, 2020.
Ambroxol is a commonly used medication in Europe, which promotes the clearance of mucus and eases coughing. It is used for the treatment for respiratory diseases, and also has anti-inflammatory properties.
Preclinical experiments suggest that ambroxol may help by bolstering cellular waste disposal systems. In PD, there is evidence to suggest that abnormal proteins are accumulated in cells and are not being disposed of properly. In particular, researchers have found that ambroxol increases levels of a protein called GCase in cells. By increasing levels of GCase, ambroxol allows cells to remove waste more effectively. This would ideally keep cells healthier for longer and therefore slow down the progression of PD.
This study has been funded by CPT in partnership with Van Andel Institute (VAI) and the John Black Charitable Foundation. Further studies will be required to determine if ambroxol is having a disease-modifying impact. CPT, VAI and the John Black Charitable Foundation are now actively exploring the next steps in the clinical testing of ambroxol.
Sign up to receive our email newsletter with links to the latest content. ACNR is free, thanks to the support of advertisers. The editorial content is peer reviewed and remains completely independent unless clearly specified.