Press release on behalf of Veriton Pharma Ltd
Published online: 2/10/17
Veriton Pharma Ltd is announced as the new company name for Special Products Ltd.
This name change has occurred both to incorporate the company’s past heritage with its future ambitions to make trusted medicines available for everyday living.
The Chief Commercial Officer commented:
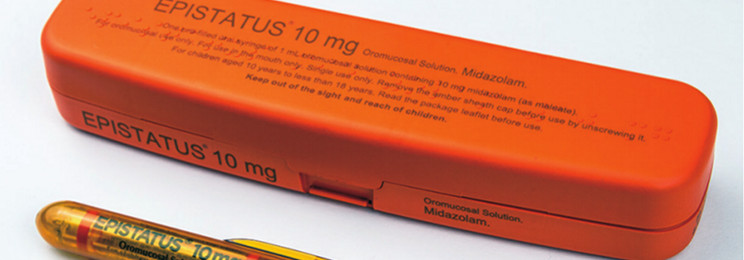
With the launch of Epistatus® 10mg in 1mL Oromucosal Solution Midazolam (as maleate), on the 8th of September 2017, and our extensive range of existing unlicensed medicines, we would like to assure our customers and partners that the excellent service and quality they have come to expect from us as a company will continue and all contacts at the company will remain the same.
With its legacy of technical expertise, which began in a world-renowned children’s hospital over 20 years ago, Veriton Pharma sources and supplies licensed medicines for CNS and over 75 high-quality, UK batch manufactured unlicensed medicines in the fields of epilepsy, neurology and rare paediatric conditions that licensed products cannot meet.
Headquartered in Weybridge, UK, Veriton Pharma, is a privately owned, speciality pharmaceutical company, which also has regional offices in the Middle East and Australia.
For further information about Veriton Pharma, visit www.veritonpharma.com
Epistatus Prescribing Information
EPISTATUS® 10mg oromucosal solution midazolam (as maleate). Please consult Summary of Product Characteristics before prescribing.
Presentation & composition: oromucosal solution. Each 1mL of solution contains 10mg of midazolam (as maleate). Excipients with a known effect: ethanol 197mg/mL, liquid maltitol 675mg.
Indication: Treatment of prolonged, acute, convulsive seizures in children and adolescents aged 10 to less than 18 years. Epistatus must only be used by parents / caregivers where the patient has been diagnosed to have epilepsy.
Dosage: For children and adolescents aged 10 to less than 18 years the standard dose is 10 mg (1.0 mL). Carers should only administer a single dose. If the seizure has not stopped within 10 minutes after administration, emergency medical assistance must be sought. Patients should be kept under supervision by a carer who remains with the patient. A second or repeat dose when seizures re-occur after an initial response should not be given without prior medical advice.
Administration: For oromucosal use only. Using the pre-filled oral syringe provided, administer, over a period of 2-3 seconds, approximately half of the prescribed dose to each buccal cavity. For detailed instructions please refer to the Summary of Product Characteristics.
Contra-indications: Hypersensitivity to midazolam, benzodiazepines or to any of the excipients. Myasthenia gravis; severe respiratory insufficiency; sleep apnoea syndrome; severe hepatic impairment.
Warnings & Precautions: Caution in patients with chronic respiratory insufficiency (may further depress respiration). For oromucosal use only. Take care to avoid the risk of choking. Midazolam should be used with caution in patients with chronic renal failure or impaired hepatic function (may accumulate); or cardiac function (may decrease clearance). Debilitated patients are more prone to the central nervous system (CNS) effects of benzodiazepines and, therefore, lower doses may be required. Midazolam should be avoided in patients with a medical history of alcohol or drug abuse. May cause anterograde amnesia. Contains maltitol and ethanol.
Interactions: Please consult the Summary of Product Characteristics for full details. Midazolam is metabolized by cytochrome P450 3A4 isozyme (CYP3A4). Inhibitors and inducers of CYP3A4 may increase and decrease the plasma concentration respectively. In the presence of CYP3A4 inhibition the duration of effect of a single dose of oromucosal midazolam may be prolonged; careful clinical monitoring is recommended. Midazolam may interact with other hepatically metabolized medicinal products. Co-administration with other sedative / hypnotic agents and CNS depressants, including alcohol, is likely to result in enhanced sedation and respiratory depression. Additional alcohol intake should be strongly avoided.
Pregnancy and lactation: Midazolam may be used during pregnancy if clearly necessary. The risk for new-born infants should be considered in the event of administration in the third trimester. Midazolam passes in low quantities into breast milk (0.6%); it may not be necessary to stop breast-feeding following a single dose.
Driving and machines: midazolam has a major influence on the ability to drive or use machines. The patient should be warned not to drive or use machines until fully recovered.
Side effects: Respiratory depression occurs at a rate of up to 5% although this is a known complication of convulsive seizures as well as being related to benzodiazepine use. Common: sedation, somnolence, depressed level of consciousness, respiratory depression, nausea & vomiting. Uncommon: pruritus, rash, urticaria. Following injection, additional adverse reactions have very rarely been reported (including respiratory arrest and cardiac arrest); these may be of relevance to oromucosal administration. Consult the Summary of Product Characteristics before prescribing.
Legal classification: POM
NHS Price: 10mg in 1mL prefilled syringe – £45.76
Marketing authorisation number: PL 16786/0003
Marketing authorisation holder: Veriton Pharma Limited, Unit 16, Trade City, Avro Way, Brooklands Business Park, Weybridge, Surrey, KT13 0YF, United Kingdom.
Date of last revision: July 2017.
EDM/1036/2017
Date of preparation August 2017